

Roles and mechanisms of cellular senescence in regulation of tissue homeostasis
- PMID: 23360516
- PMCID: PMC7657106
- DOI: 10.1111/cas.12118
Roles and mechanisms of cellular senescence in regulation of tissue homeostasis
Abstract
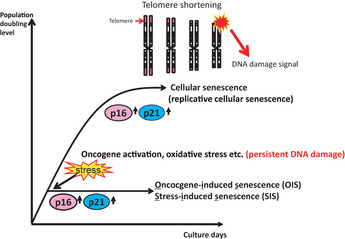
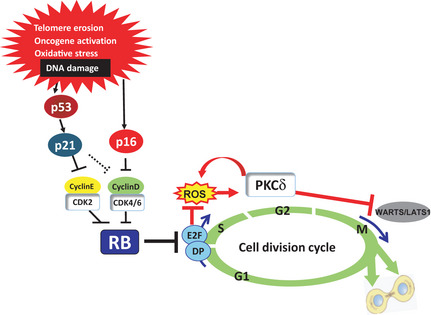
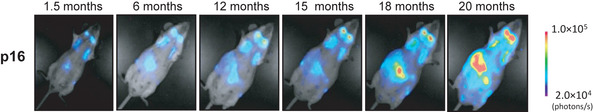
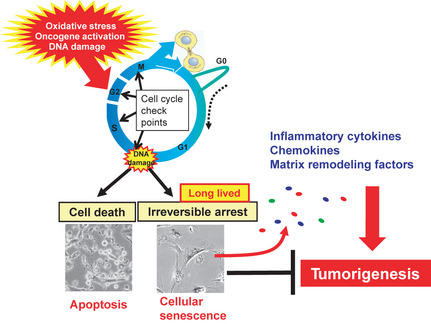
Cellular senescence is the state of irreversible cell cycle arrest that can be induced by a variety of potentially oncogenic stimuli and has therefore long been considered to suppress tumorigenesis, acting as a guardian of homeostasis. However, surprisingly, emerging evidence reveals that senescent cells also promote secretion of a series of inflammatory cytokines, chemokines, growth factors and matrix remodeling factors, which alter the local tissue environment and contribute to chronic inflammation and cancer. This newly identified senescence phenotype, termed the senescence-associated secretory phenotype (SASP) or the senescence-messaging secretome (SMS), is induced by DNA damage that promotes the induction of cellular senescence. All of these senescence-associated secreting factors are involved in homeostatic disorders such as cancer. Therefore, it is quite possible that accumulation of senescent cells during the aging process in vivo might contribute to age-related increases in homeostatic disorders. In this review, current knowledge of the molecular and cellular biology of cellular senescence is introduced, focusing on its positive and negative roles in controlling tissue homeostasis in vivo.
© 2013 Japanese Cancer Association.
Figures
References
-
- Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature 2007; 448: 767–74. - PubMed
-
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120: 513–22. - PubMed
-
- Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech Ageing Dev 2006; 127: 16–24. - PubMed
-
- Sharpless NE, DePinho RA. Cancer: crime and punishment. Nature 2005; 436: 636–7. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
