

Inferring the kinetics of stochastic gene expression from single-cell RNA-sequencing data
- PMID: 23360624
- PMCID: PMC3663116
- DOI: 10.1186/gb-2013-14-1-r7
Inferring the kinetics of stochastic gene expression from single-cell RNA-sequencing data
Abstract
Background: Genetically identical populations of cells grown in the same environmental condition show substantial variability in gene expression profiles. Although single-cell RNA-seq provides an opportunity to explore this phenomenon, statistical methods need to be developed to interpret the variability of gene expression counts.
Results: We develop a statistical framework for studying the kinetics of stochastic gene expression from single-cell RNA-seq data. By applying our model to a single-cell RNA-seq dataset generated by profiling mouse embryonic stem cells, we find that the inferred kinetic parameters are consistent with RNA polymerase II binding and chromatin modifications. Our results suggest that histone modifications affect transcriptional bursting by modulating both burst size and frequency. Furthermore, we show that our model can be used to identify genes with slow promoter kinetics, which are important for probabilistic differentiation of embryonic stem cells.
Conclusions: We conclude that the proposed statistical model provides a flexible and efficient way to investigate the kinetics of transcription.
Figures
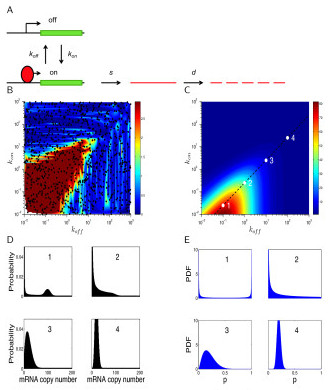
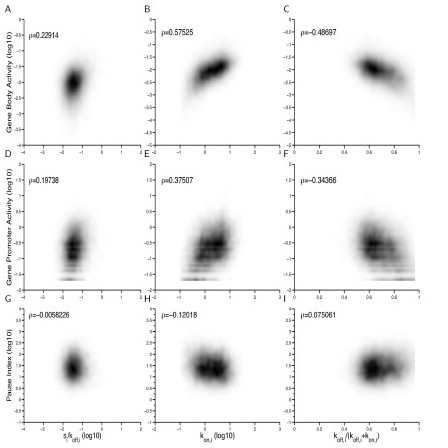
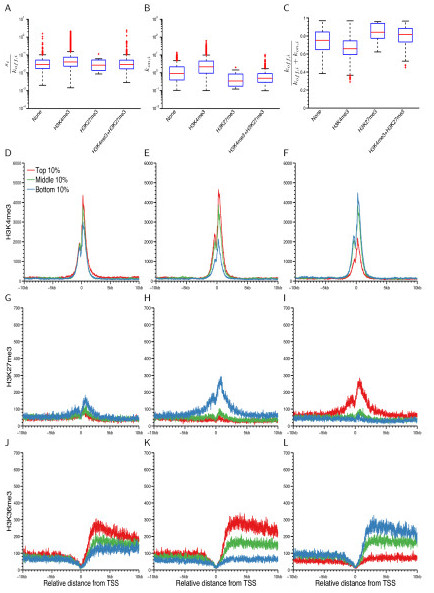
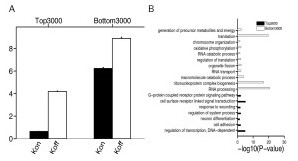
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
