

Inhibition of G-protein-coupled receptor kinase 2 (GRK2) triggers the growth-promoting mitogen-activated protein kinase (MAPK) pathway
- PMID: 23362259
- PMCID: PMC3597814
- DOI: 10.1074/jbc.M112.428078
Inhibition of G-protein-coupled receptor kinase 2 (GRK2) triggers the growth-promoting mitogen-activated protein kinase (MAPK) pathway
Abstract
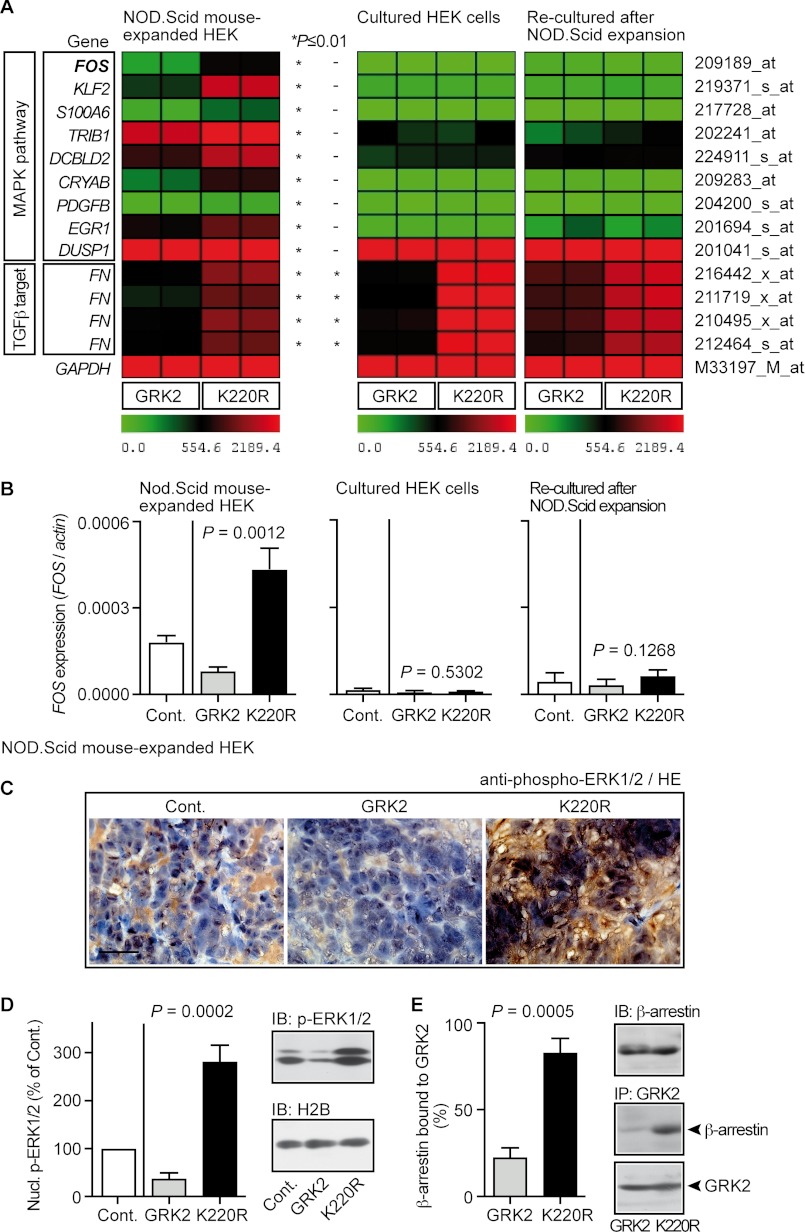
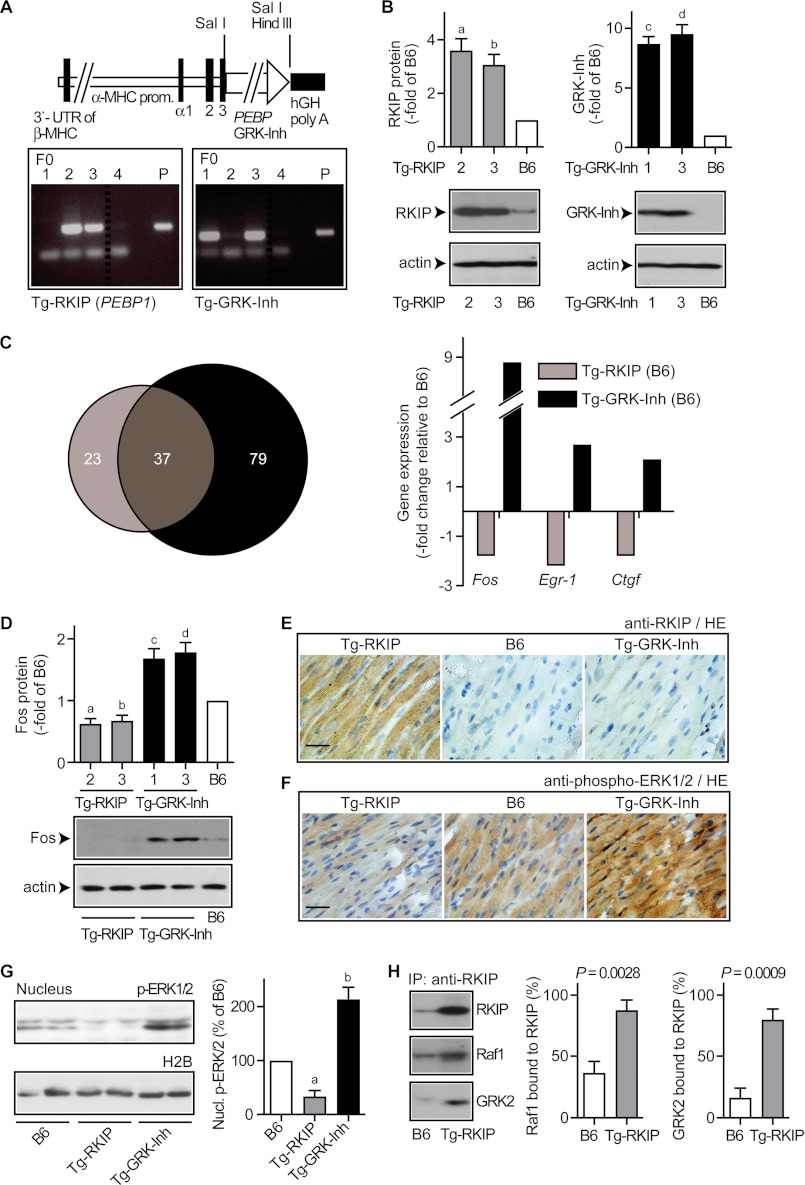
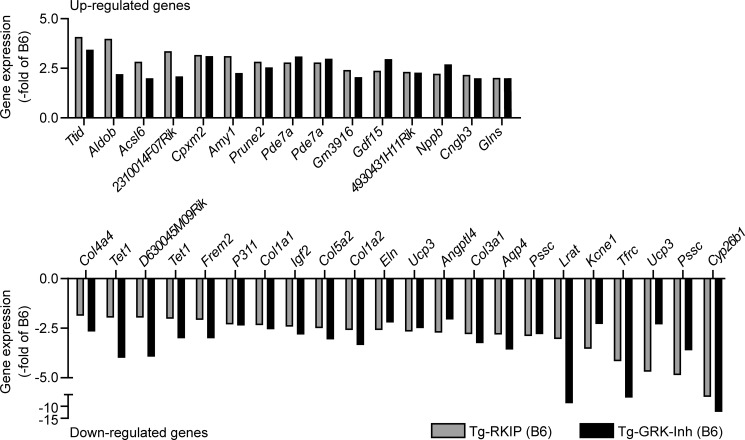
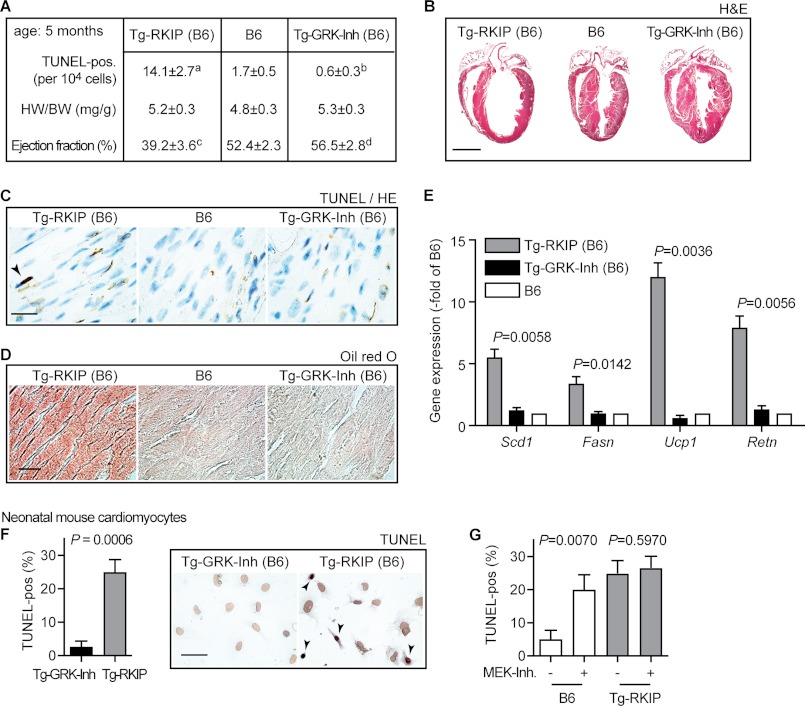
Inhibition of G-protein-coupled receptor kinase 2 (GRK2) is an emerging treatment option for heart failure. Because GRK2 is also indispensable for growth and development, we analyzed the impact of GRK2 inhibition on cell growth and proliferation. Inhibition of GRK2 by the dominant-negative GRK2-K220R did not affect the proliferation of cultured cells. In contrast, upon xenograft transplantation of cells into immunodeficient mice, the dominant-negative GRK2-K220R or a GRK2-specific peptide inhibitor increased tumor mass. The enhanced tumor growth upon GRK2 inhibition was attributed to the growth-promoting MAPK pathway because dual inhibition of the GRK2 and RAF-MAPK axis by the Raf kinase inhibitor protein (RKIP) did not increase tumor mass. The MAPK cascade contributed to the cardioprotective profile of GRK2 inhibition by preventing cardiomyocyte death, whereas dual inhibition of RAF/MAPK and GRK2 by RKIP induced cardiomyocyte apoptosis, cardiac dysfunction, and signs of heart failure. Thus, cardioprotective signaling induced by GRK2 inhibition is overlapping with tumor growth promotion.
Figures


Similar articles
-
The RAF Kinase Inhibitor Protein (RKIP): Good as Tumour Suppressor, Bad for the Heart.Cells. 2022 Feb 14;11(4):654. doi: 10.3390/cells11040654. Cells. 2022. PMID: 35203304 Free PMC article. Review.
-
Inhibition of G-protein-coupled Receptor Kinase 2 Prevents the Dysfunctional Cardiac Substrate Metabolism in Fatty Acid Synthase Transgenic Mice.J Biol Chem. 2016 Feb 5;291(6):2583-600. doi: 10.1074/jbc.M115.702688. Epub 2015 Dec 15. J Biol Chem. 2016. PMID: 26670611 Free PMC article.
-
Sensitization of the Angiotensin II AT1 Receptor Contributes to RKIP-Induced Symptoms of Heart Failure.Front Med (Lausanne). 2019 Jan 9;5:359. doi: 10.3389/fmed.2018.00359. eCollection 2018. Front Med (Lausanne). 2019. PMID: 30687708 Free PMC article.
-
G protein-coupled receptor kinase 2 (GRK2) is a Rho-activated scaffold protein for the ERK MAP kinase cascade.Cell Signal. 2013 Dec;25(12):2831-9. doi: 10.1016/j.cellsig.2013.08.031. Epub 2013 Sep 7. Cell Signal. 2013. PMID: 24018045
-
GRK2-dependent desensitization downstream of G proteins.J Recept Signal Transduct Res. 2008;28(1-2):59-70. doi: 10.1080/10799890801941939. J Recept Signal Transduct Res. 2008. PMID: 18437630 Review.
Cited by
-
NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases.Int J Mol Sci. 2019 Mar 30;20(7):1599. doi: 10.3390/ijms20071599. Int J Mol Sci. 2019. PMID: 30935055 Free PMC article. Review.
-
Dual role of GRK5 in cancer development and progression.Transl Med UniSa. 2016 May 16;14:28-37. eCollection 2016 May. Transl Med UniSa. 2016. PMID: 27326393 Free PMC article.
-
Restricting mitochondrial GRK2 post-ischemia confers cardioprotection by reducing myocyte death and maintaining glucose oxidation.Sci Signal. 2018 Dec 11;11(560):eaau0144. doi: 10.1126/scisignal.aau0144. Sci Signal. 2018. PMID: 30538174 Free PMC article.
-
The RAF Kinase Inhibitor Protein (RKIP): Good as Tumour Suppressor, Bad for the Heart.Cells. 2022 Feb 14;11(4):654. doi: 10.3390/cells11040654. Cells. 2022. PMID: 35203304 Free PMC article. Review.
-
Raf kinase inhibitor protein mediates myocardial fibrosis under conditions of enhanced myocardial oxidative stress.Basic Res Cardiol. 2018 Sep 6;113(6):42. doi: 10.1007/s00395-018-0700-3. Basic Res Cardiol. 2018. PMID: 30191336 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
