

Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria
- PMID: 23364466
- PMCID: PMC3646094
- DOI: 10.2119/molmed.2012.00340
Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria
Abstract
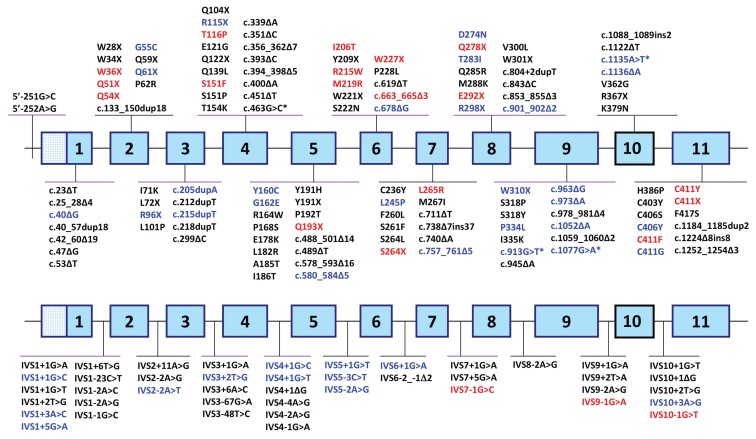
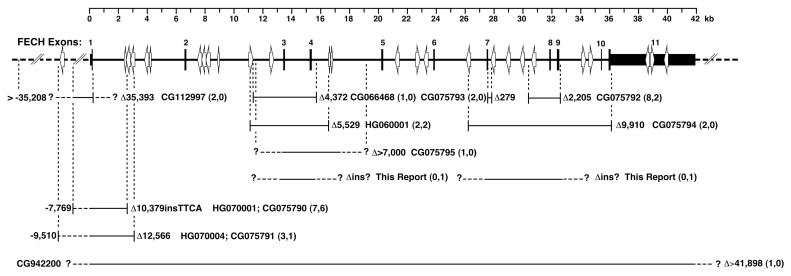
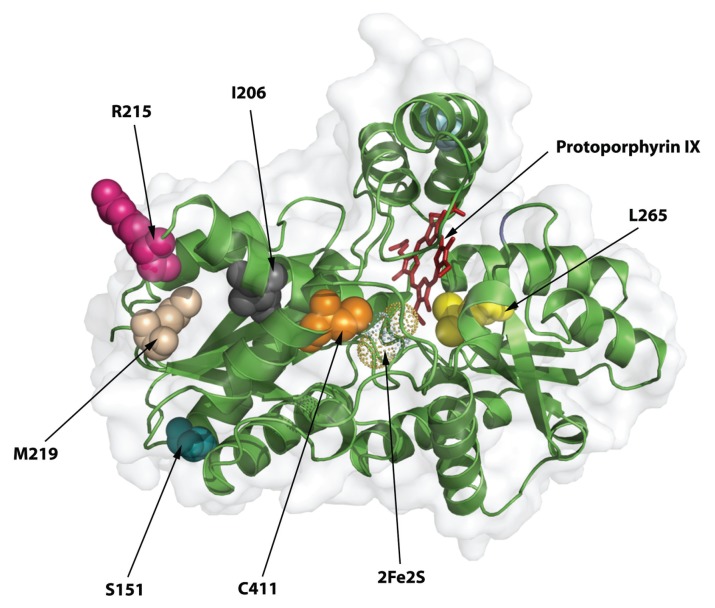
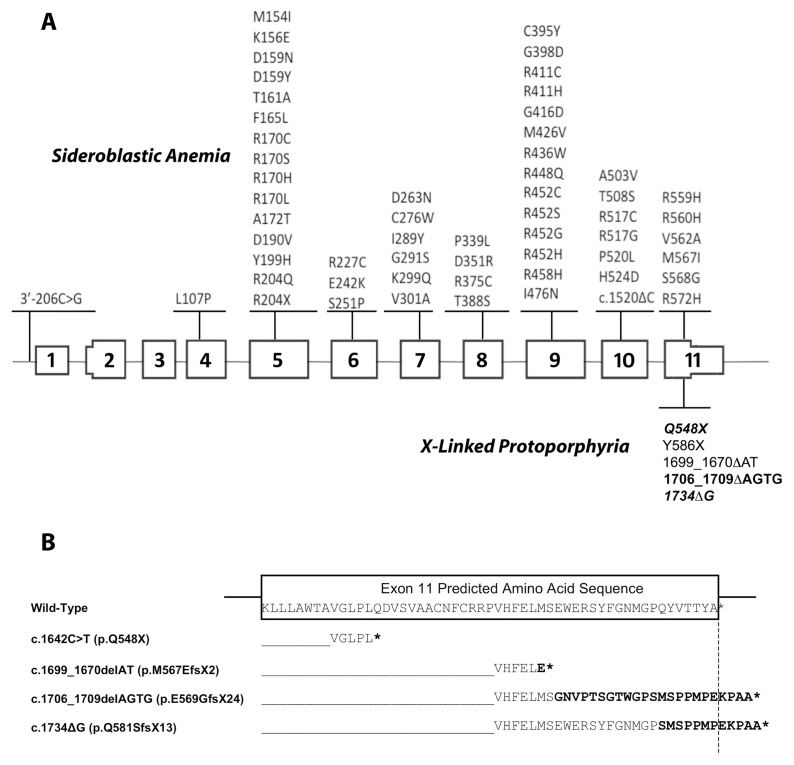
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are inborn errors of heme biosynthesis with the same phenotype but resulting from autosomal recessive loss-of-function mutations in the ferrochelatase (FECH) gene and gain-of-function mutations in the X-linked erythroid-specific 5-aminolevulinate synthase (ALAS2) gene, respectively. The EPP phenotype is characterized by acute, painful, cutaneous photosensitivity and elevated erythrocyte protoporphyrin levels. We report the FECH and ALAS2 mutations in 155 unrelated North American patients with the EPP phenotype. FECH sequencing and dosage analyses identified 140 patients with EPP: 134 with one loss-of-function allele and the common IVS3-48T>C low expression allele, three with two loss-of-function mutations and three with one loss-of-function mutation and two low expression alleles. There were 48 previously reported and 23 novel FECH mutations. The remaining 15 probands had ALAS2 gain-of-function mutations causing XLP: 13 with the previously reported deletion, c.1706_1709delAGTG, and two with novel mutations, c.1734delG and c.1642C>T(p.Q548X). Notably, XLP represented ~10% of EPP phenotype patients in North America, two to five times more than in Western Europe. XLP males had twofold higher erythrocyte protoporphyrin levels than EPP patients, predisposing to more severe photosensitivity and liver disease. Identification of XLP patients permits accurate diagnosis and counseling of at-risk relatives and asymptomatic heterozygotes.
Figures
References
-
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease; 8th edition; New York: McGraw-Hill; 2001. pp. 2991–3062.
-
- Todd DJ. Erythropoietic protoporphyria. Br. J. Dermatol. 1994;131:751–766. - PubMed
-
- Anderson KE, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005;142:439–50. - PubMed
-
- Gross U, Frank M, Doss MO. Hepatic complications of erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 1998;14:52–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
