

From natural to artificial photosynthesis
- PMID: 23365193
- PMCID: PMC3627107
- DOI: 10.1098/rsif.2012.0984
From natural to artificial photosynthesis
Abstract
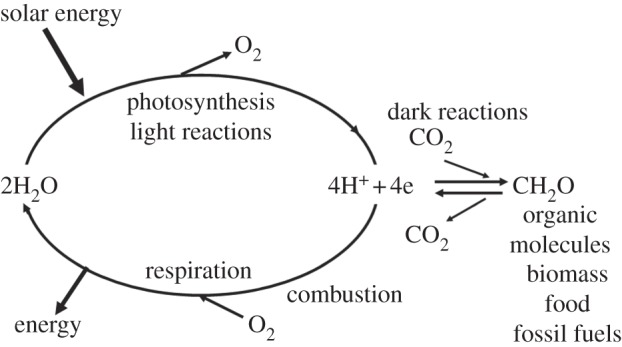
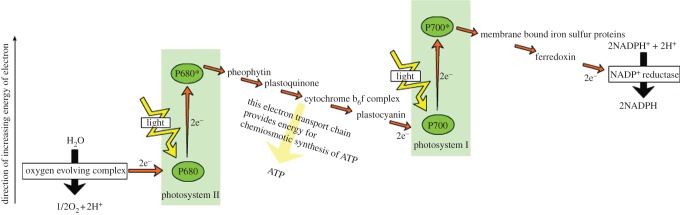
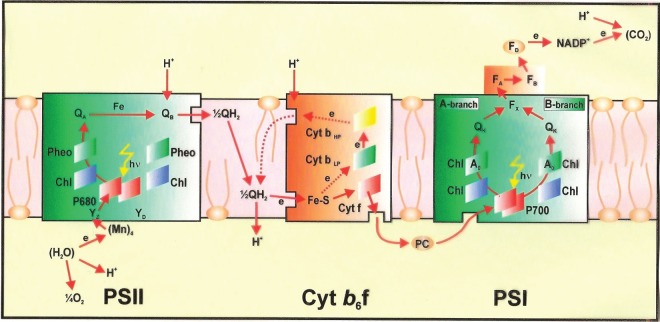
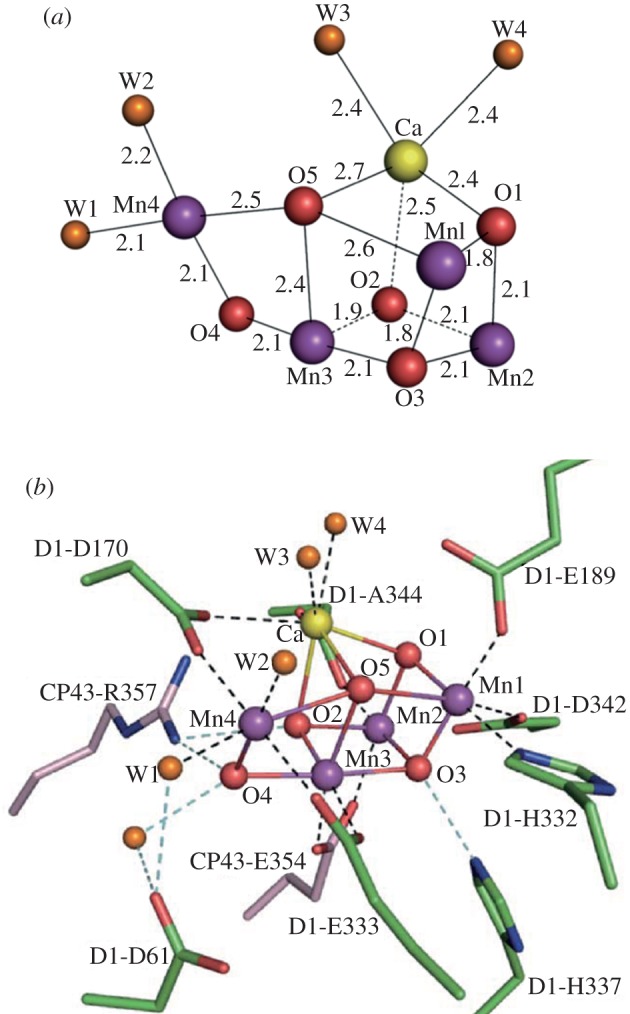
Demand for energy is projected to increase at least twofold by mid-century relative to the present global consumption because of predicted population and economic growth. This demand could be met, in principle, from fossil energy resources, particularly coal. However, the cumulative nature of carbon dioxide (CO(2)) emissions demands that stabilizing the atmospheric CO(2) levels to just twice their pre-anthropogenic values by mid-century will be extremely challenging, requiring invention, development and deployment of schemes for carbon-neutral energy production on a scale commensurate with, or larger than, the entire present-day energy supply from all sources combined. Among renewable and exploitable energy resources, nuclear fusion energy or solar energy are by far the largest. However, in both cases, technological breakthroughs are required with nuclear fusion being very difficult, if not impossible on the scale required. On the other hand, 1 h of sunlight falling on our planet is equivalent to all the energy consumed by humans in an entire year. If solar energy is to be a major primary energy source, then it must be stored and despatched on demand to the end user. An especially attractive approach is to store solar energy in the form of chemical bonds as occurs in natural photosynthesis. However, a technology is needed which has a year-round average conversion efficiency significantly higher than currently available by natural photosynthesis so as to reduce land-area requirements and to be independent of food production. Therefore, the scientific challenge is to construct an 'artificial leaf' able to efficiently capture and convert solar energy and then store it in the form of chemical bonds of a high-energy density fuel such as hydrogen while at the same time producing oxygen from water. Realistically, the efficiency target for such a technology must be 10 per cent or better. Here, we review the molecular details of the energy capturing reactions of natural photosynthesis, particularly the water-splitting reaction of photosystem II and the hydrogen-generating reaction of hydrogenases. We then follow on to describe how these two reactions are being mimicked in physico-chemical-based catalytic or electrocatalytic systems with the challenge of creating a large-scale robust and efficient artificial leaf technology.
Figures
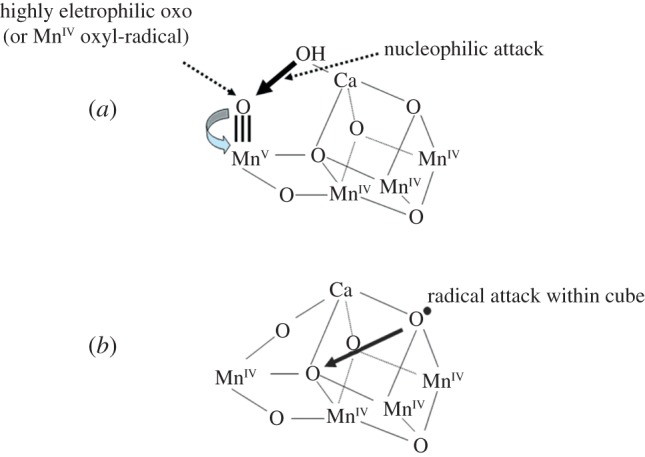
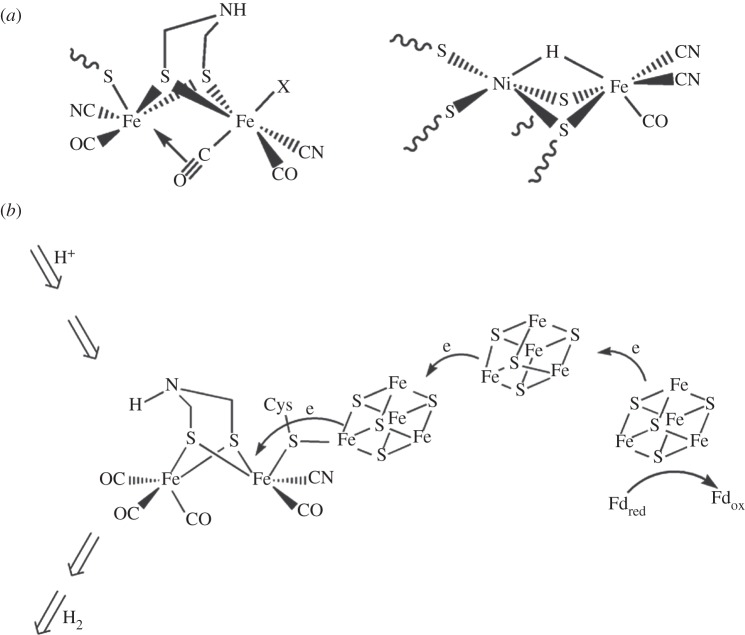
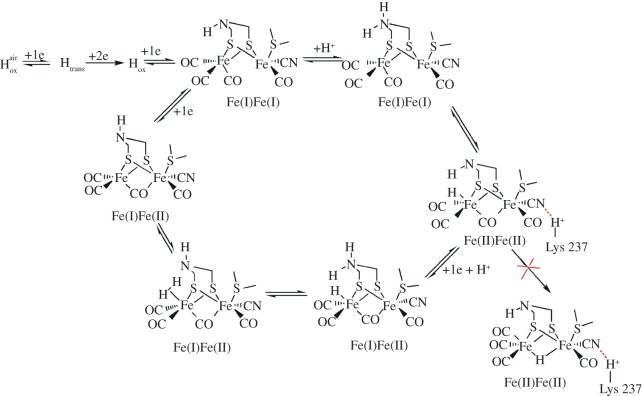
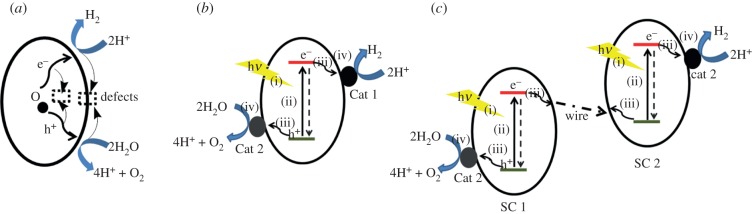
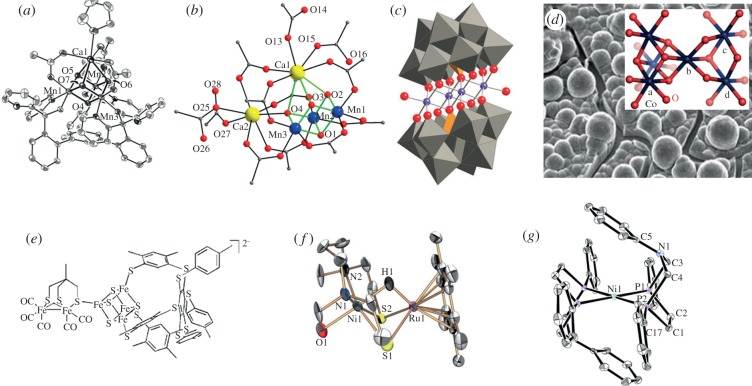
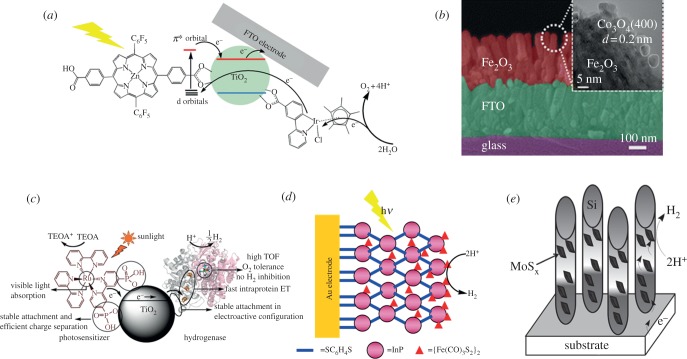
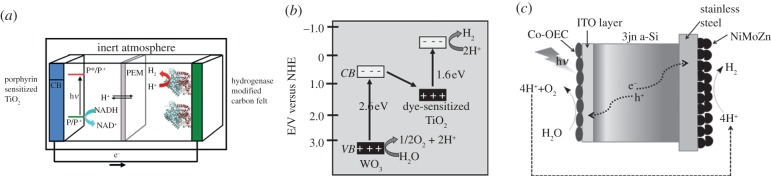
References
-
- International Energy Agency. 2012. Key World Energy Statistics 2012. Paris, France: International Energy Agency. (http://www.iea.org. )
-
- Hoffert MT, et al. 1998. Energy implications of future stabilization of atmospheric CO2 content. Nature 395, 881–88410.1038/27638 (doi:10.1038/27638) - DOI - DOI
-
- Nakicenovic N, Swart R. 2000. Special report on emissions scenarios, pp. 48–55 Washington, DC: Intergovernmental Panel on Climate Change
-
- Lewis NS, Nocera DG. 2006. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. USA 103, 15 729–15 73510.1073/pnas.0603395103 (doi:10.1073/pnas.0603395103) - DOI - DOI - PMC - PubMed
-
- United Nations Development Program 2003. World energy assessment report: energy and the challenge of sustainability. New York, NY: United Nations
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
