

Smoke extract impairs adenosine wound healing: implications of smoke-generated reactive oxygen species
- PMID: 23371060
- PMCID: PMC3707376
- DOI: 10.1165/rcmb.2011-0273OC
Smoke extract impairs adenosine wound healing: implications of smoke-generated reactive oxygen species
Abstract
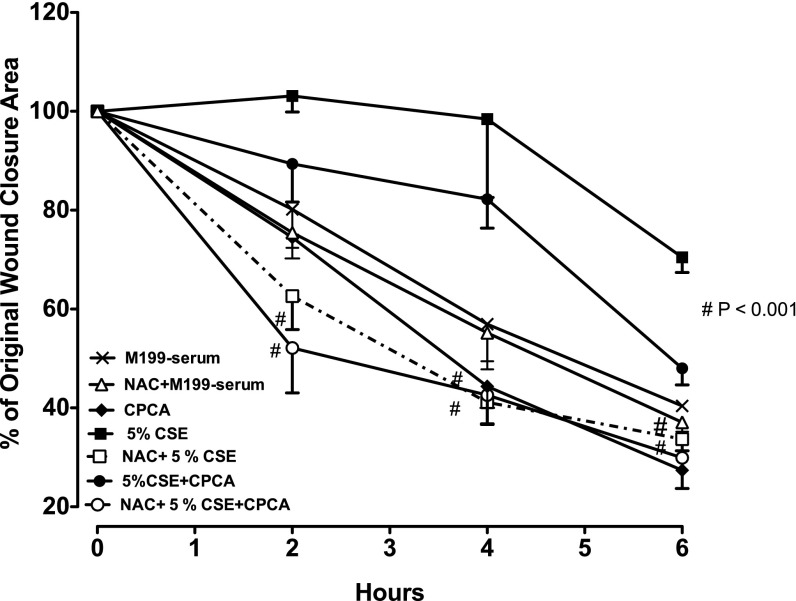
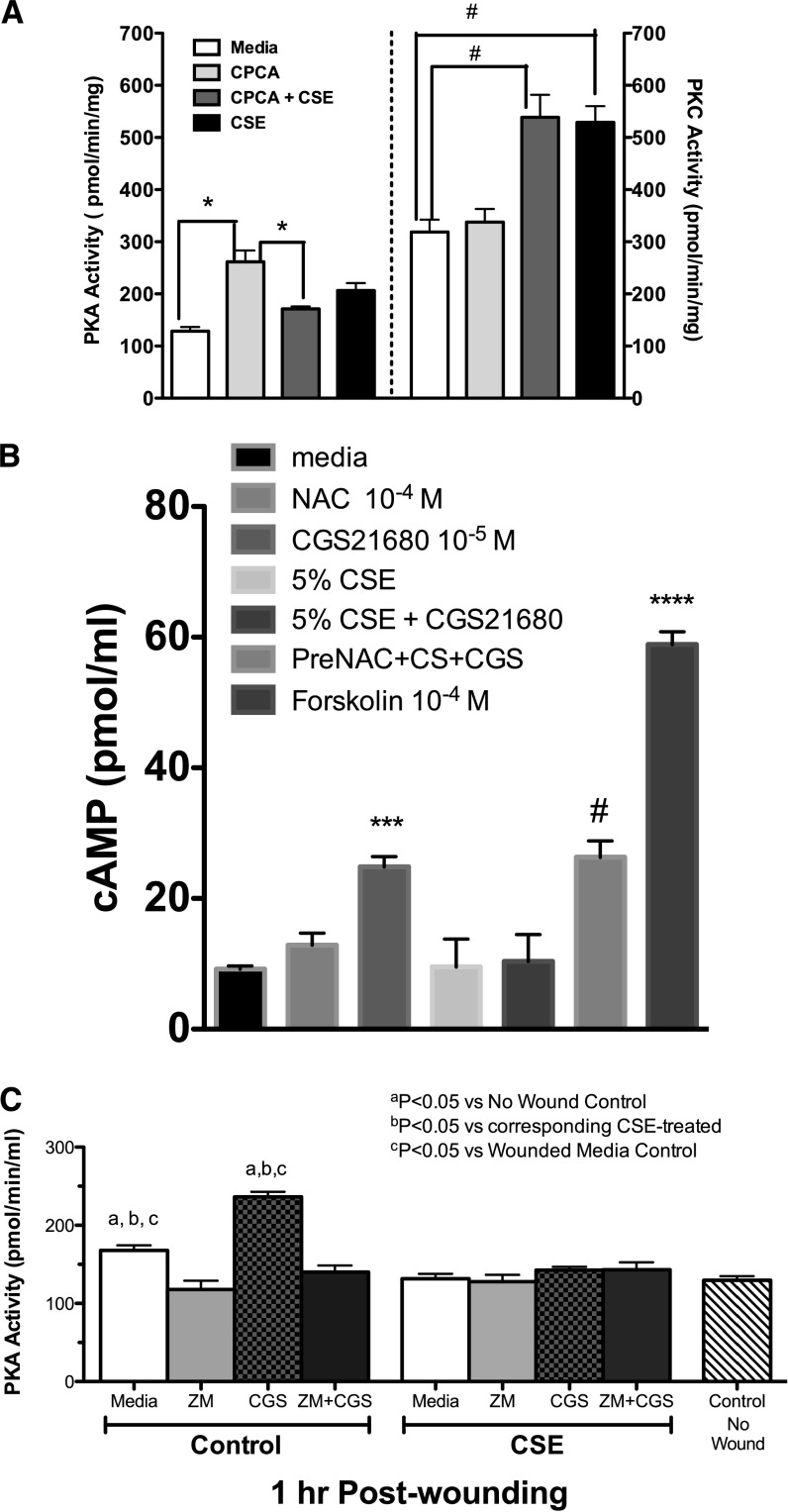
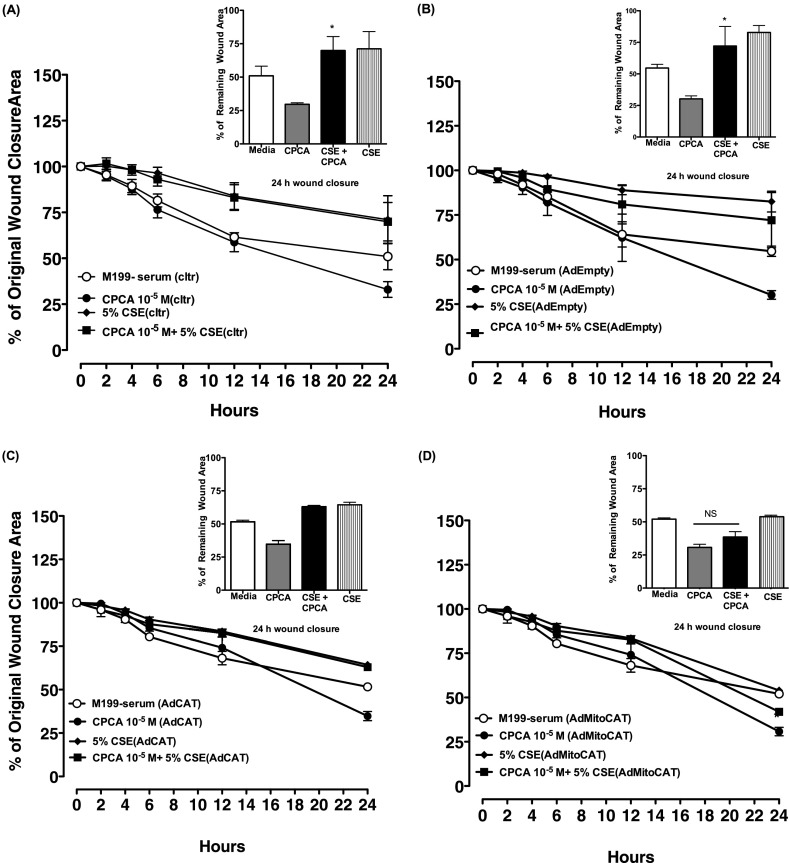
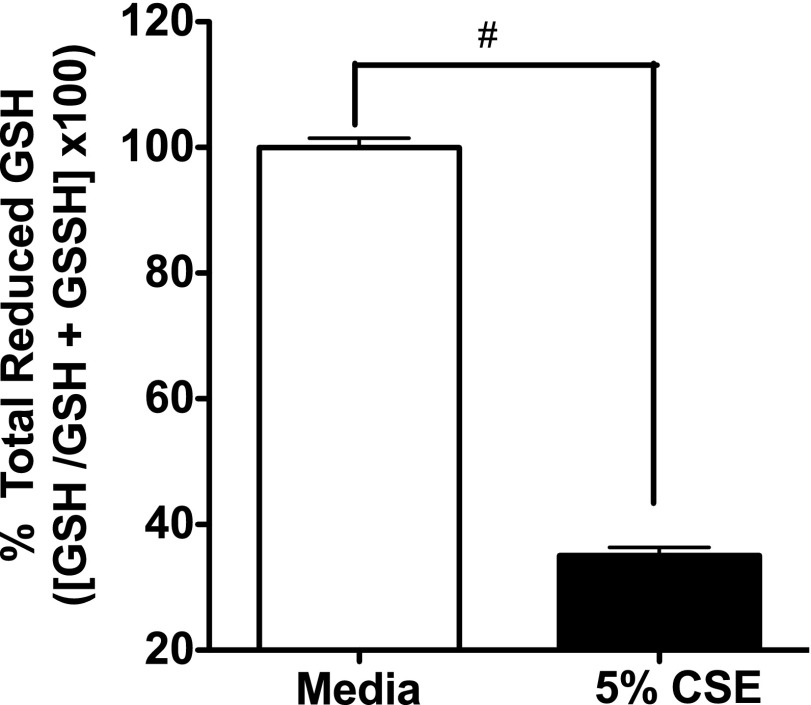
Adenosine concentrations are elevated in the lungs of patients with asthma and chronic obstructive pulmonary disease, where it balances between tissue repair and excessive airway remodeling. We previously demonstrated that the activation of the adenosine A2A receptor promotes epithelial wound closure. However, the mechanism by which adenosine-mediated wound healing occurs after cigarette smoke exposure has not been investigated. The present study investigates whether cigarette smoke exposure alters adenosine-mediated reparative properties via its ability to induce a shift in the oxidant/antioxidant balance. Using an in vitro wounding model, bronchial epithelial cells were exposed to 5% cigarette smoke extract, were wounded, and were then stimulated with either 10 μM adenosine or the specific A2A receptor agonist, 5'-(N-cyclopropyl)-carboxamido-adenosine (CPCA; 10 μM), and assessed for wound closure. In a subset of experiments, bronchial epithelial cells were infected with adenovirus vectors encoding human superoxide dismutase and/or catalase or control vector. In the presence of 5% smoke extract, significant delay was evident in both adenosine-mediated and CPCA-mediated wound closure. However, cells pretreated with N-acetylcysteine (NAC), a nonspecific antioxidant, reversed smoke extract-mediated inhibition. We found that cells overexpressing mitochondrial catalase repealed the smoke extract inhibition of CPCA-stimulated wound closure, whereas superoxide dismutase overexpression exerted no effect. Kinase experiments revealed that smoke extract significantly reduced the A2A-mediated activation of cyclic adenosine monophosphate-dependent protein kinase. However, pretreatment with NAC reversed this effect. In conclusion, our data suggest that cigarette smoke exposure impairs A2A-stimulated wound repair via a reactive oxygen species-dependent mechanism, thereby providing a better understanding of adenosine signaling that may direct the development of pharmacological tools for the treatment of chronic inflammatory lung disorders.
Figures



References
-
- Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 2009;24:298–306. - PubMed
-
- Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. - PubMed
-
- Blackburn MR. Too much of a good thing: adenosine overload in adenosine–deaminase–deficient mice. Trends Pharmacol Sci. 2003;24:66–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
