

Diagnosing mucopolysaccharidosis IVA
- PMID: 23371450
- PMCID: PMC3590423
- DOI: 10.1007/s10545-013-9587-1
Diagnosing mucopolysaccharidosis IVA
Abstract
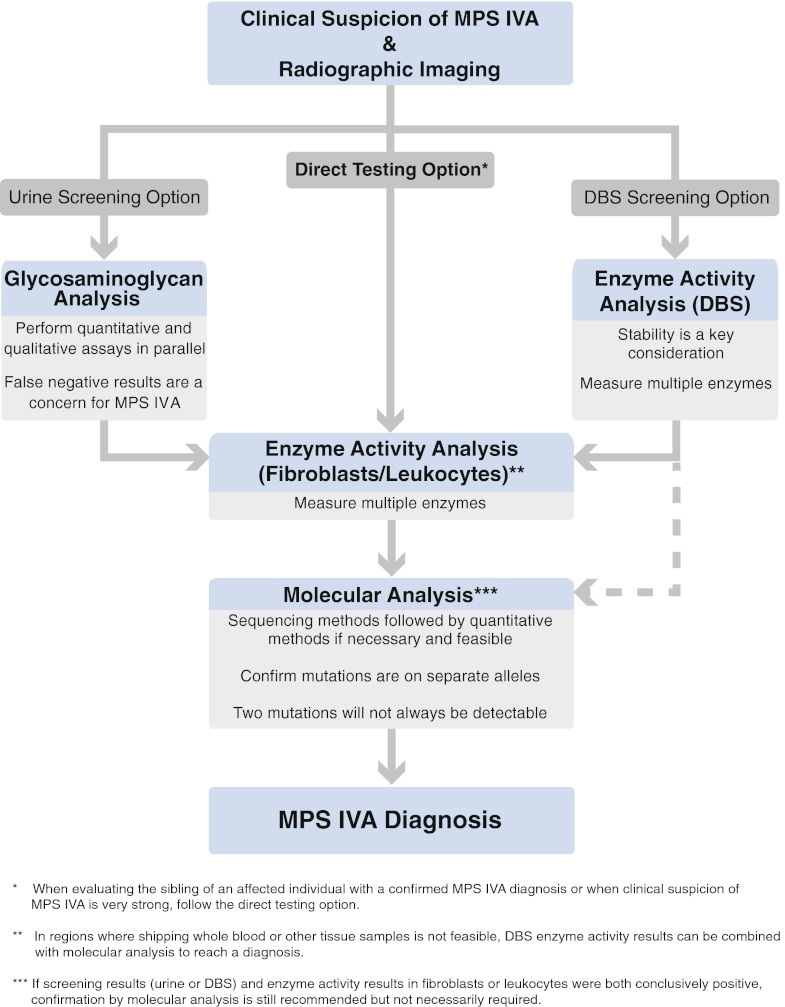
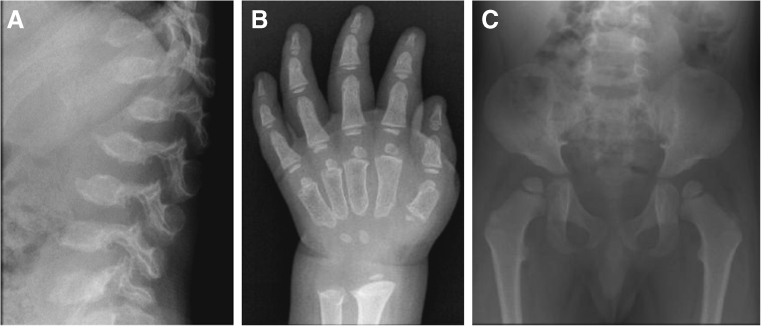
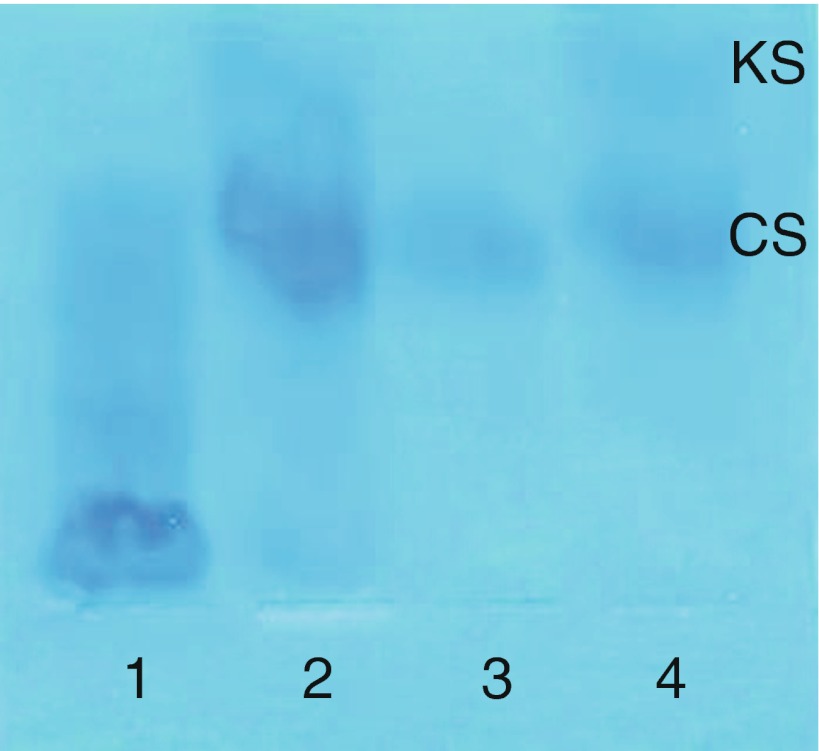
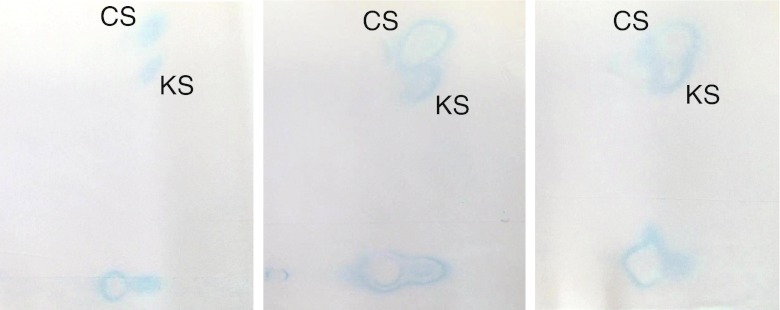
Mucopolysaccharidosis IVA (MPS IVA; Morquio A syndrome) is an autosomal recessive lysosomal storage disorder resulting from a deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS) activity. Diagnosis can be challenging and requires agreement of clinical, radiographic, and laboratory findings. A group of biochemical genetics laboratory directors and clinicians involved in the diagnosis of MPS IVA, convened by BioMarin Pharmaceutical Inc., met to develop recommendations for diagnosis. The following conclusions were reached. Due to the wide variation and subtleties of radiographic findings, imaging of multiple body regions is recommended. Urinary glycosaminoglycan analysis is particularly problematic for MPS IVA and it is strongly recommended to proceed to enzyme activity testing even if urine appears normal when there is clinical suspicion of MPS IVA. Enzyme activity testing of GALNS is essential in diagnosing MPS IVA. Additional analyses to confirm sample integrity and rule out MPS IVB, multiple sulfatase deficiency, and mucolipidoses types II/III are critical as part of enzyme activity testing. Leukocytes or cultured dermal fibroblasts are strongly recommended for enzyme activity testing to confirm screening results. Molecular testing may also be used to confirm the diagnosis in many patients. However, two known or probable causative mutations may not be identified in all cases of MPS IVA. A diagnostic testing algorithm is presented which attempts to streamline this complex testing process.
Conflict of interest statement
Authors Bainbridge, Burin, Church, D’Almeida, van Diggelen, Fietz, Harmatz, Hendriksz, Lukacs, Pasquali, and Wood received consultant fees and limited travel reimbursement from BioMarin Pharmaceutical Inc. (BioMarin) for participating in the Global MPS IVA Laboratory Diagnostics Scientific Summit in Prague on June 16th–18th, 2011. Authors Bainbridge, Burin, Chien, Church, van Diggelen, Giugliani, Harmatz, Hendriksz, Hwu, Lukacs, Pasquali, Thompson, Tylee, Wood, and Yu received consultant fees and limited travel reimbursement from BioMarin for participating in the Global MPS IVA Laboratory Diagnostics Scientific Summit in San Diego on February 6th–7th, 2012. Authors Bainbridge, Beck, Burin, Chien, D’Almeida, van Diggelen, Fietz, Giugliani, Harmatz, Hendriksz, Hwu, Ketteridge, Lukacs, Thompson, Tylee, and Wood were compensated by BioMarin for completing a survey on the diagnosis of MPS IVA. Authors Hawley and Miller are employees of BioMarin and have direct financial interest and investments in BioMarin. Authors Giugliani, Harmatz, Hendriksz, Lukacs, and Tylee have served or are serving on advisory boards for BioMarin. Authors Giugliani, Harmatz, Hendriksz, and Ketteridge are current or recent participants in BioMarin sponsored clinical trials. Authors Harmatz and Hendriksz have assisted in the design of clinical trials evaluating BioMarin products. Authors Beck, Harmatz, Hendriksz, and Lukacs have received research support from BioMarin. Authors Chien, D’Almeida, van Diggelen, Fietz, Giugliani, Harmatz, Hendriksz, Ketteridge, Thompson, Wood, and Yu have received consulting fees or other remuneration from BioMarin. Authors Church, D’Almeida, and Tylee have received travel grants from BioMarin. Authors Burin and Schenone have acted as expert witnesses on the subject of this manuscript. Through their laboratory, authors Church and Tylee provide a diagnostic service for MPS for samples being sent from Turkey which is funded by BioMarin, a dried blood spot diagnostic service for Fabry and Pompe diseases which is funded by Genzyme Corporation, and baseline lysosomal acid lipase measurement service for patients recruited into ongoing clinical trials for Wolman/CESD which is funded by Synageva BioPharma Corporation.
Figures

References
-
- Beaudet AL. Disorders of glycoprotein degradation: mannosidosis, fucosidosis, sialidosis, and aspartylglycosaminuria. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, Brown MS, editors. The metabolic basis of inheritied disease. New York: McGraw-Hill; 1983. pp. 788–802.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
