

Β-funaltrexamine inhibits chemokine (CXCL10) expression in normal human astrocytes
- PMID: 23376103
- PMCID: PMC3604055
- DOI: 10.1016/j.neuint.2013.01.013
Β-funaltrexamine inhibits chemokine (CXCL10) expression in normal human astrocytes
Abstract
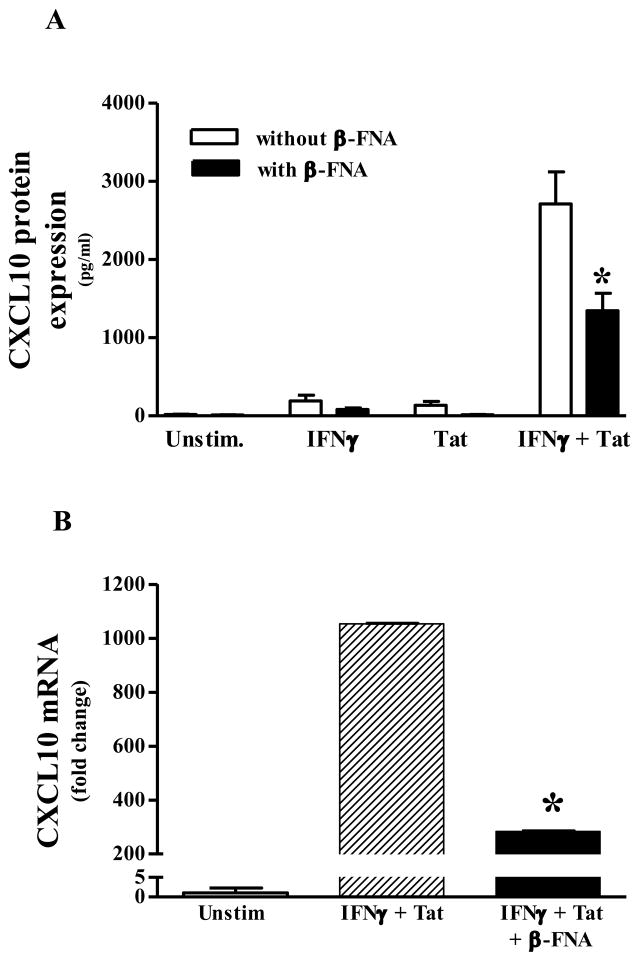
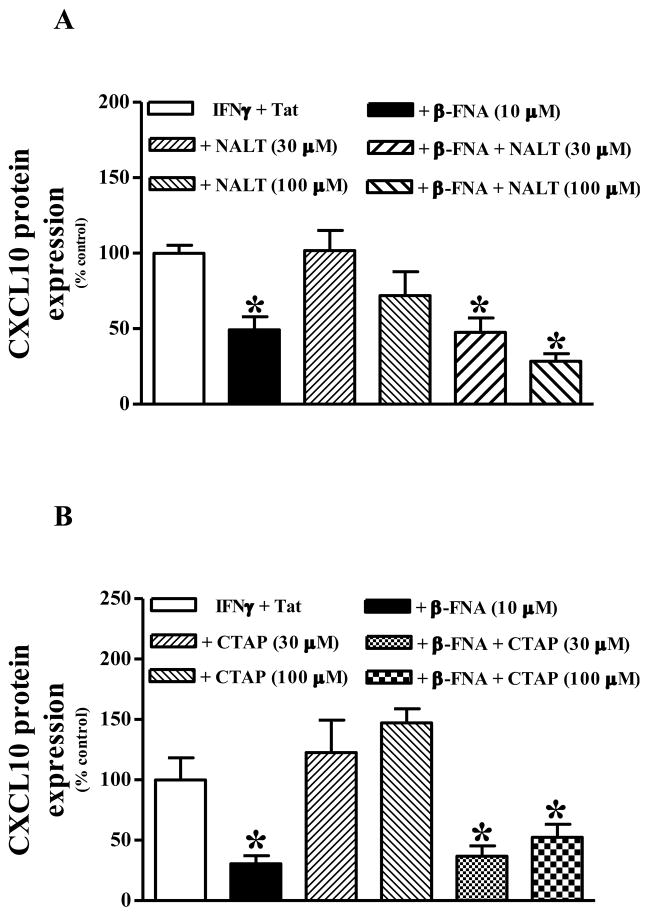
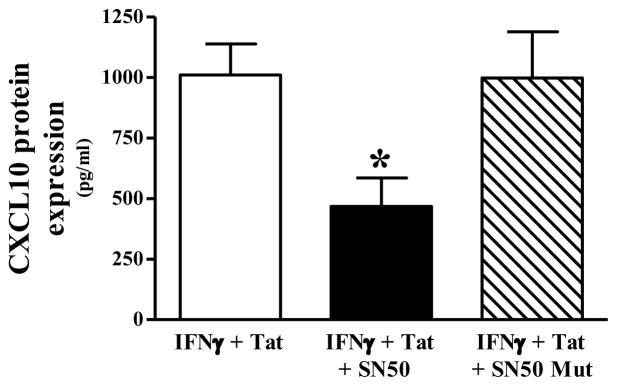
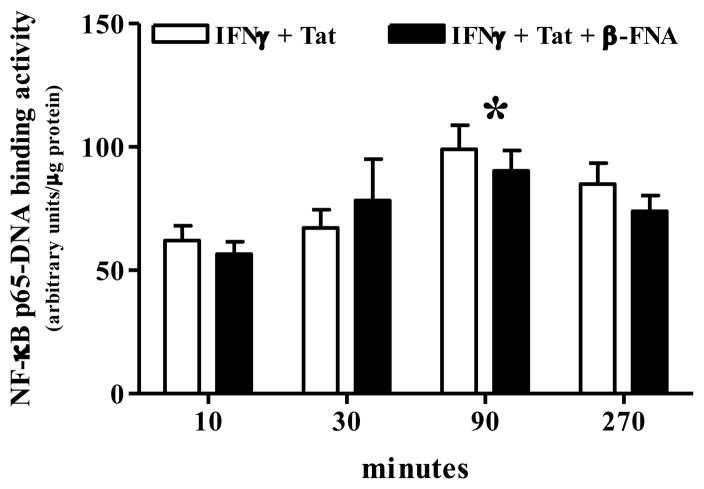
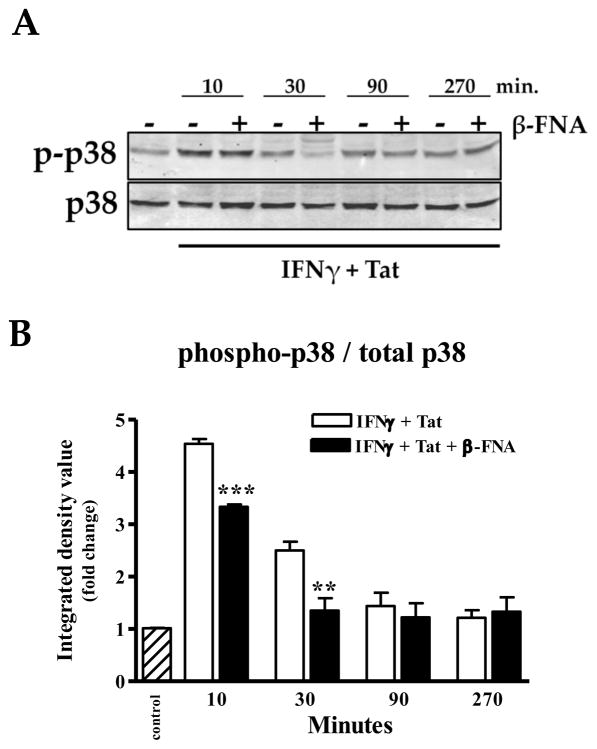
Neuroinflammation is an integral component of neurodegenerative disorders, CNS infection and trauma. Astroglial chemokines, such as CXCL10, are instrumental in neuroinflammatory signaling as well as neurotoxicity. We have utilized proinflammatory-induced CXCL10 expression in normal human astrocytes (NHA) as a model in which to assess the anti-inflammatory actions of the selective, mu-opioid receptor (MOR) antagonist, β-funaltrexamine (β-FNA). Interferon (IFN)γ+HIV-1 Tat-induced CXCL10 expression (secreted protein and mRNA) was inhibited by co-treatment with β-FNA. Neither the MOR-selective antagonist, D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2 (CTAP) nor the nonselective opioid receptor antagonist, naltrexone inhibited IFNγ+HIV-1 Tat-induced CXCL10 expression. Furthermore, co-treatment with excess CTAP or naltrexone did not prevent β-FNA mediated inhibition of IFNγ+HIV-1 Tat-induced CXCL10 expression. Additionally, we utilized an inhibitor of NF-κB activation (SN50) to demonstrate that IFNγ+HIV-1 Tat-induced CXCL10 expression is NF-κB-dependent in NHA. Subsequent experiments revealed that β-FNA did not significantly affect NF-κB activation. Interestingly, we discovered that β-FNA inhibited p38 activation as indicated by decreased expression of phospho-p38. Together, these findings suggest that the inhibitory actions of β-FNA are MOR-independent and mediated, in part, via a transcriptional mechanism. These findings add to our understanding of the mechanism by which chemokine expression is inhibited by β-FNA. In conjunction with future investigations, these novel findings are expected to provide insights into the development of safe and effective treatments for neuroinflammation.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Anti-inflammatory actions of β-funaltrexamine in a mouse model of lipopolysaccharide-induced inflammation.Inflammopharmacology. 2023 Feb;31(1):349-358. doi: 10.1007/s10787-022-01113-9. Epub 2022 Dec 17. Inflammopharmacology. 2023. PMID: 36527567
-
The opioid antagonist, β-funaltrexamine, inhibits NF-κB signaling and chemokine expression in human astrocytes and in mice.Eur J Pharmacol. 2015 Sep 5;762:193-201. doi: 10.1016/j.ejphar.2015.05.040. Epub 2015 May 22. Eur J Pharmacol. 2015. PMID: 26007645 Free PMC article.
-
The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells.J Neuroimmunol. 2007 May;186(1-2):141-9. doi: 10.1016/j.jneuroim.2007.03.021. Epub 2007 May 1. J Neuroimmunol. 2007. PMID: 17475341 Free PMC article.
-
HIV-1 Tat co-operates with IFN-gamma and TNF-alpha to increase CXCL10 in human astrocytes.PLoS One. 2009 May 28;4(5):e5709. doi: 10.1371/journal.pone.0005709. PLoS One. 2009. PMID: 19479051 Free PMC article.
-
The opioid antagonist, β-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice.Physiol Behav. 2017 May 1;173:52-60. doi: 10.1016/j.physbeh.2017.01.037. Epub 2017 Jan 24. Physiol Behav. 2017. PMID: 28130086
Cited by
-
The effects of chronic, continuous β-funaltrexamine pre-treatment on lipopolysaccharide-induced inflammation and behavioral deficits in C57BL/6J mice.J Inflamm (Lond). 2024 Sep 2;21(1):33. doi: 10.1186/s12950-024-00407-9. J Inflamm (Lond). 2024. PMID: 39223594 Free PMC article.
-
Anti-inflammatory effects of β-FNA are sex-dependent in a pre-clinical model of LPS-induced inflammation.J Inflamm (Lond). 2023 Jan 25;20(1):4. doi: 10.1186/s12950-023-00328-z. J Inflamm (Lond). 2023. PMID: 36698151 Free PMC article.
-
β-Funaltrexamine Displayed Anti-inflammatory and Neuroprotective Effects in Cells and Rat Model of Stroke.Int J Mol Sci. 2020 May 29;21(11):3866. doi: 10.3390/ijms21113866. Int J Mol Sci. 2020. PMID: 32485857 Free PMC article.
-
Anti-inflammatory actions of β-funaltrexamine in a mouse model of lipopolysaccharide-induced inflammation.Inflammopharmacology. 2023 Feb;31(1):349-358. doi: 10.1007/s10787-022-01113-9. Epub 2022 Dec 17. Inflammopharmacology. 2023. PMID: 36527567
-
The opioid antagonist, β-funaltrexamine, inhibits NF-κB signaling and chemokine expression in human astrocytes and in mice.Eur J Pharmacol. 2015 Sep 5;762:193-201. doi: 10.1016/j.ejphar.2015.05.040. Epub 2015 May 22. Eur J Pharmacol. 2015. PMID: 26007645 Free PMC article.
References
-
- Agrawal L, Louboutin JP, Strayer DS. Preventing HIV-1 tat-induced neuronal apoptosis using antioxidant enzymes: Mechanistic and therapeutic implications. Virology. 2007;363:462–472. - PubMed
-
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. - PubMed
-
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. - PubMed
-
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
