

Review
doi: 10.1101/cshperspect.a009514.
Cystic fibrosis transmembrane conductance regulator (ABCC7) structure
Affiliations
- PMID: 23378596
- PMCID: PMC3552343
- DOI: 10.1101/cshperspect.a009514
Item in Clipboard
Review
Cystic fibrosis transmembrane conductance regulator (ABCC7) structure
Cold Spring Harb Perspect Med.
.
Abstract
Structural studies of the cystic fibrosis transmembrane conductance regulator (CFTR) are reviewed. Like many membrane proteins, full-length CFTR has proven to be difficult to express and purify, hence much of the structural data available is for the more tractable, independently expressed soluble domains. Therefore, this chapter covers structural data for individual CFTR domains in addition to the sparser data available for the full-length protein. To set the context for these studies, we will start by reviewing structural information on model proteins from the ATP-binding cassette (ABC) transporter superfamily, to which CFTR belongs.
Figures
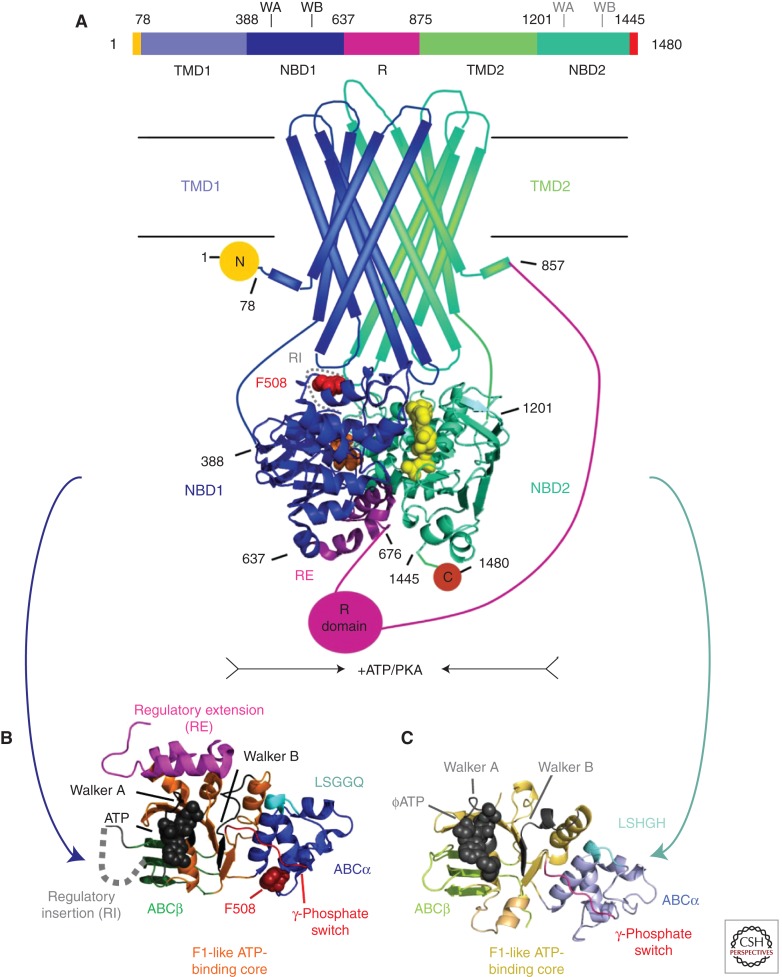
Domain organization of CFTR and simplified consensus gating model. (A) The three-dimensional domain organization of CFTR is schematized below a bar showing its linear sequence organization. NBD stands for nucleotide-binding domain, TMD for transmembrane domain, R for regulatory domain, RI for the regulatory insertion in NBD1, RE for the regulatory extension at the junction between NBD1 and the R domain, and N and C for the amino- and carboxy-terminal cytoplasmic tails, respectively. ATP is shown in orange (bound to the Walker motifs in NBD1) or yellow (bound to the Walker motifs in NBD2) space-filling representation. The NBDs are represented by ribbon diagrams of crystal structures of the isolated human domains (PDB IDs 2BBO for hNBD1 and 3GD7 for hNBD2), which are shown interacting in an ATP-sandwich heterodimer conformation modeled based on least-squares alignment to the crystal structure of the low-affinity homodimer of hNBD1-Δ(RI,RE) (PDB ID 2PZE). PKA stands for protein kinase A, which uses ATP to phosphorylate and thereby activate the CFTR channel; phosphorylation presumably occurs primarily on residues in the R domain. (B) Ribbon diagram showing the subdomain organization of hNBD1 (PDB ID 2BBO), with the F1-like ATP-binding core subdomain shown in orange, the ABCβ subdomain in dark green, the ABCα subdomain in blue, the γ-phosphate switch in light red, the LSGGQ signature sequence in light cyan, the RE in magenta, the RI in gray (depicted as a dotted line), and Mg-ATP in black space-filling representation. Residue F508 (dark red) and its Walker A and B motifs are labeled. (C) Ribbon diagram showing the subdomain organization of hNBD2 (PDB ID 3GD7), with the F1-like ATP-binding core subdomain shown in orange, the ABCβ subdomain in light green, the ABCα subdomain in gray-blue, the γ-phosphate switch in magenta, the LSHGH signature sequence in dark cyan, and Mg-ϕATP (phenyl-ATP) in black space-filling representation. Its Walker A and B motifs are labeled. (D) Schematic diagrams illustrating a simplified version of the accepted model for the mechanochemistry of CFTR channel gating. The upper row shows a top view looking down on the NBDs from the cytoplasm, whereas the lower row shows a side view of the transmembrane structure (with the phospholipid bilayer schematized as two parallel black lines). The coloring is equivalently to panel A except for ATP, which is shown here in red. The opening of the transmembrane chloride channel in CFTR is gated by mechanical reorientation of the NBDs upon ATP binding at their interface, which drives formation of an ATP-sandwich heterodimer structure. Phosphorylation of the R domain by PKA is believed to facilitate channel opening at least in part by disrupting binding interactions between polypeptide segments in the R domain and surfaces in the NBDs mediating tight interdomain structural interactions in the open conformation of the channel.
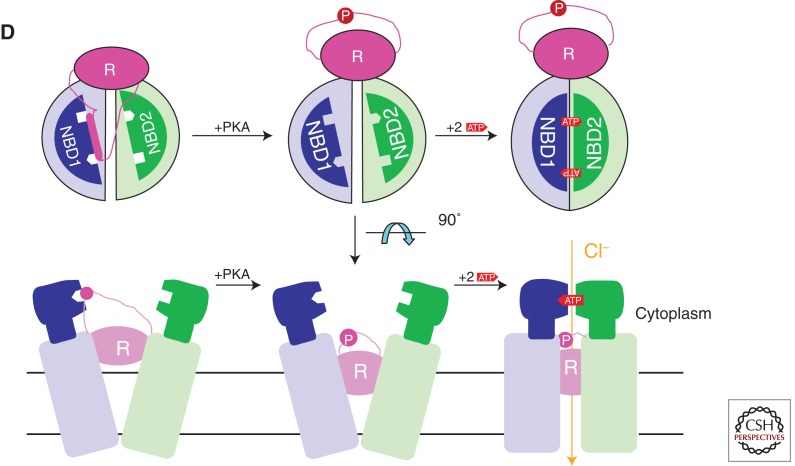
Domain organization of CFTR and simplified consensus gating model. (A) The three-dimensional domain organization of CFTR is schematized below a bar showing its linear sequence organization. NBD stands for nucleotide-binding domain, TMD for transmembrane domain, R for regulatory domain, RI for the regulatory insertion in NBD1, RE for the regulatory extension at the junction between NBD1 and the R domain, and N and C for the amino- and carboxy-terminal cytoplasmic tails, respectively. ATP is shown in orange (bound to the Walker motifs in NBD1) or yellow (bound to the Walker motifs in NBD2) space-filling representation. The NBDs are represented by ribbon diagrams of crystal structures of the isolated human domains (PDB IDs 2BBO for hNBD1 and 3GD7 for hNBD2), which are shown interacting in an ATP-sandwich heterodimer conformation modeled based on least-squares alignment to the crystal structure of the low-affinity homodimer of hNBD1-Δ(RI,RE) (PDB ID 2PZE). PKA stands for protein kinase A, which uses ATP to phosphorylate and thereby activate the CFTR channel; phosphorylation presumably occurs primarily on residues in the R domain. (B) Ribbon diagram showing the subdomain organization of hNBD1 (PDB ID 2BBO), with the F1-like ATP-binding core subdomain shown in orange, the ABCβ subdomain in dark green, the ABCα subdomain in blue, the γ-phosphate switch in light red, the LSGGQ signature sequence in light cyan, the RE in magenta, the RI in gray (depicted as a dotted line), and Mg-ATP in black space-filling representation. Residue F508 (dark red) and its Walker A and B motifs are labeled. (C) Ribbon diagram showing the subdomain organization of hNBD2 (PDB ID 3GD7), with the F1-like ATP-binding core subdomain shown in orange, the ABCβ subdomain in light green, the ABCα subdomain in gray-blue, the γ-phosphate switch in magenta, the LSHGH signature sequence in dark cyan, and Mg-ϕATP (phenyl-ATP) in black space-filling representation. Its Walker A and B motifs are labeled. (D) Schematic diagrams illustrating a simplified version of the accepted model for the mechanochemistry of CFTR channel gating. The upper row shows a top view looking down on the NBDs from the cytoplasm, whereas the lower row shows a side view of the transmembrane structure (with the phospholipid bilayer schematized as two parallel black lines). The coloring is equivalently to panel A except for ATP, which is shown here in red. The opening of the transmembrane chloride channel in CFTR is gated by mechanical reorientation of the NBDs upon ATP binding at their interface, which drives formation of an ATP-sandwich heterodimer structure. Phosphorylation of the R domain by PKA is believed to facilitate channel opening at least in part by disrupting binding interactions between polypeptide segments in the R domain and surfaces in the NBDs mediating tight interdomain structural interactions in the open conformation of the channel.
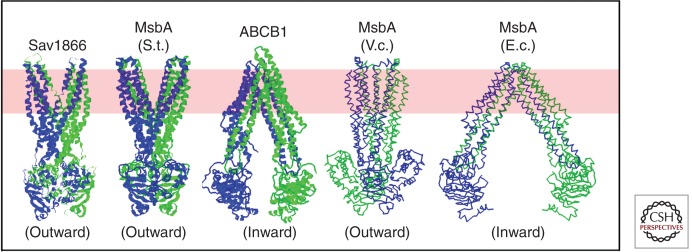
Published crystal structures of ABC transporters homologous to CFTR (Dawson and Locher 2006, 2007; Ward et al. 2007; Aller et al. 2009). Ribbon diagrams are shown for three structures in which sidechains were modeled (left), whereas backbone traces are shown for the two structures in which the limited resolution of the diffraction data prevented side-chain assignment (right) (Ward et al. 2007). Structures have been determined for the MsbA orthologs from Salmonella typhimurium (S.t.), Vibrio cholera (V.c.), and Escherichia coli (E.c). All structures shown are homodimers, with the exception of ABCB1 (also called P-glycoprotein or Pgp), which has two NBDs and two transmembrane domains fused in a single polypeptide chain. The two subunits in each homodimer are colored differently (i.e., blue vs. green), as are the amino-terminal and carboxy-terminal halves of ABCB1. The approximate location of the lipid bilayer is illustrated by a pink band. The SAV1866, MsbA (S.t.), and MsbA (V.c.) structures are in outward-facing conformations, whereas the ABCB1 and MsbA (E.c.) structures are in inward-facing conformations.
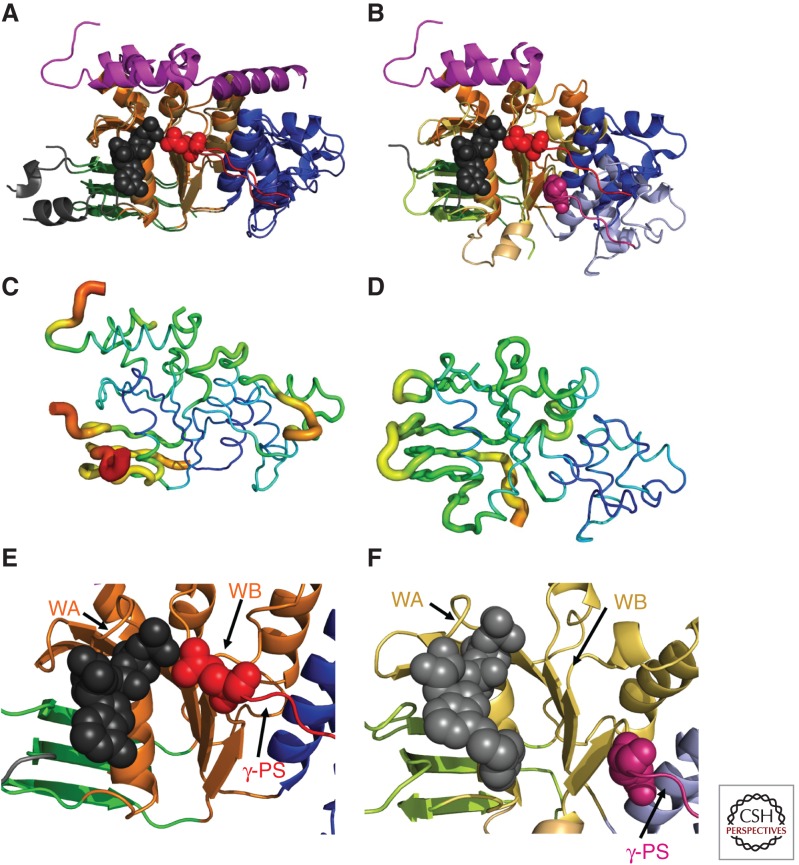
Subdomain reorientation and backbone B-factors in hNBD1 and hNBD2 crystal structures. All panels show the hNBD1 (panels A, C, and E on the left and panel B on the right) and hNBD2 (panels B, D, and F on the right) domains in the same orientation after least-squares alignment of the β-strands in their ATP-binding cores (i.e., the ABCβ subdomain plus the F1-like core subdomain). The ribbon diagrams in panels A,B and E,F are colored equivalently to Fig. 1B,C (i.e., orange or yellow for the F1-like ATP-binding core subdomain, shades of blue for the ABCα subdomain, shades of green for the ABCβ subdomain, and red or magenta for the γ-phosphate switch or Q-loop). The bound ATP molecules (black or gray) and the invariant glutamines in the γ-phosphate switch (Q493 in hNBD1 in red and Q1291 in hNBD2 in magenta) are shown in space-filling representation. (A,B) Least-squares superpositions of two full-length F508-hNBD1 structures (panel A—PDB IDs 2BBO and 1 XMI) and of one of these with hNBD2 (panel B—PDB IDs 2BBO and 3GD7, respectively). The 7° rotation of the ABCα subdomain between the two hNBD1 structures shown here represents the largest displacement observed when comparing any two hNBD1 crystal structure, all of which have ATP bound in the active site. As discussed in the text, the 17° rotation and ∼10 Å displacement of the ABCα subdomain in hNBD2 relative to the ATP-bound conformation in hNBD1 likely prevents formation of a hydrolytically active ATP-sandwich heterodimer with hNBD1 in vitro, even though extensive evidence supports formation of such a complex coupled to opening of the CFTR channel in vivo (as schematized in Fig. 1D) (Gadsby 2004; Mense et al. 2006; Aleksandrov et al. 2008, 2009; Hwang et al. 2009). (C,D) Equivalent views of the hNBD1 (panel C) and hNBD2 (panel D) structures from panel B with the thickness and color of the backbone trace representing their mean backbone B-factors. B-factors quantify the variation or disorder in atomic position in a crystal structure, with larger B-factors indicating more disorder. The default encoding scheme in PyMOL was used to generate these images (i.e., 0.6–4.0 Å backbone thickness and a linear blue-to-green-to-red color ramp for B-factors running from 32–80 Å2 for hNBD1 and from 19–76 Å2 for hNBD2). (E,F) Close-up views of the ATP-binding sites in the crystal structures of F508-hNBD1-Δ(RI,RE) (panel E – PDB ID 2PZE) and hNBD2 (panel F – PDB ID 3GD7). Labels indicate the locations of the Walker A (WA) and Walker B (WB) motifs in the F1-like core subdomain and the γ-phosphate switch (γ-PS) linking the core subdomain to the amino terminus of the ABCα subdomain. Although hNBD1 is catalytically inactive, due at least in part to the substitution of a serine for the catalytic glutamate residue adjacent to the Walker B motif, its γ-PS adopts a catalytically active conformation in which its amino-terminal glutamine (Q493, red spheres) contacts the Mg2+ cofactor of ATP, as observed in ATP-bound structures of catalytically active NBDs from a wide variety of ABC transporters (Hung et al. 1998; Smith et al. 2002). In contrast, the γ-PS in hNBD2 adopts a different conformation resulting in an ∼17 Å displacement of the amide group of its amino-terminal glutamine (Q1291, magenta spheres) from the ATP molecule bound in the active site.
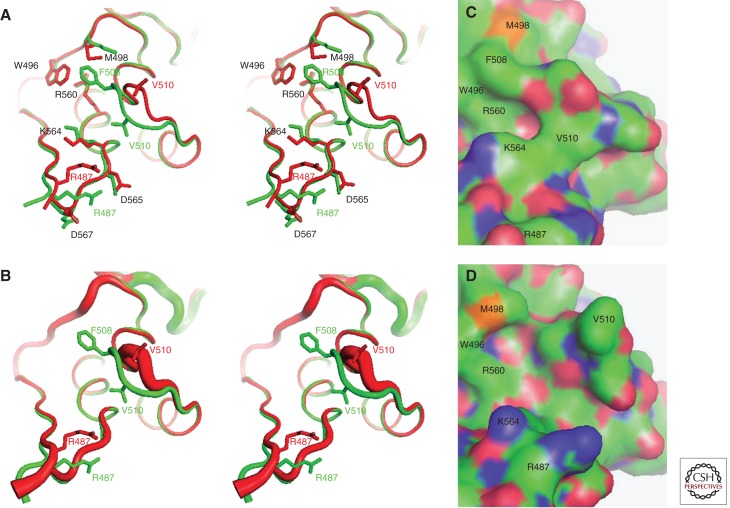
Structural consequences of the F508del mutation in hNBD1. Structures of hNBD1 with and without the F508del mutation were aligned based on least-squares superposition of the three α-helices comprising the core of the ABCα subdomain. (A) Stereopair showing the interface between the F1-like core subdomain and the ABCα subdomain in F508 (green, PDB ID 2BBO) and F508del (red, PDB ID 1XMJ) hNBD1 structures. Selected sidechains are shown in ball-and-stick representation. The site of attachment of hNBD1 to the transmembrane domains of CFTR is proximal to the viewer in this orientation, and the γ-PS is at the upper left (i.e., the backbone segment including residues W496 and M498). (B) Stereopair showing the same view of the same hNBD1 structures using the same color scheme, but with the thickness of the backbone traces encoding their mean backbone B-factors (using the default scheme in PyMOL, with 0.6–4.0 Å radius representing 33–77 Å2 and 12–69 Å2 for the F508 and F508del structures, respectively). (C,D) Surface representations of the same structures showing the alterations in local surface topography caused by the F508del mutation. These two panels show the same view of the molecular surfaces of F508 (panel C) and F508del (panel D) hNBD1, with carbon atoms colored green, oxygen atoms colored red, nitrogen atoms colored blue, and sulfur atoms colored orange. Note that the structures shown here contain seven point mutations included in hNBD1 constructs because of their beneficial influence on yield during purification—F409L, F429S, F433L, G550E, R553Q, R555K, and H667R. Extensive analyses led to the conclusion that these mutations do not significantly perturb the native ground-state conformation of hNBD1 (Lewis et al. 2010) but that they do stabilize it thermodynamically compared to an aggregation-prone molten globule intermediate that accounts for both the instability of F508del-CFTR in vivo and the progressive loss of hNBD1 constructs in vitro during purification (Protasevich et al. 2010; Wang et al. 2010). (The images shown here are from Lewis et al. 2010; adapted, with permission, from the authors.)
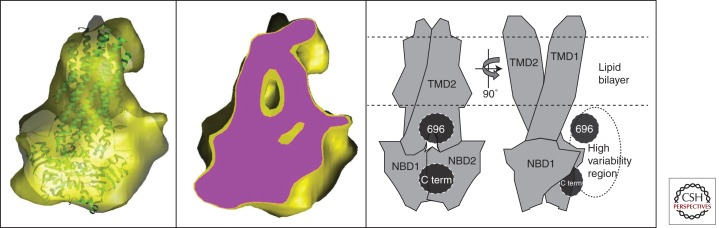
Cryo electron microscopy (EM) analysis of human CFTR. The yellow surface in the left panel shows structural data (Coulomb density map) derived from single-particle EM analysis of CFTR purified in the nonionic detergent β-dodecylmaltoside (β-DDM) but eluted from the final chromatography column in the charged detergent lyso-phosphatidylglycerol (l-PG) just above its critical micelle concentration (cmc) (Zhang et al. 2009). Similar images are obtained from samples eluted from the final column in β-DDM (Zhang et al. 2009). The crystal structure of Sav1866 (green ribbon trace) has been docked into the map. The center panel shows a slice through the same map (purple surface), revealing its internal features. The right panel shows two orthogonal views of a model for the domain organization of CFTR derived from the cryo-EM map shown in the other panels. The locations of the carboxyl terminus (C term) and the region around residue 696 in the R region were inferred from immuno-gold-labeling of poly-Histidine tags inserted into CFTR at these positions (Zhang et al. 2010). A high variability region revealed by cryo-EM (Zhang et al. 2009) is hypothesised to represent the R region.
References
-
- Aleksandrov AA, Aleksandrov L, Riordan JR 2002a. Nucleoside triphosphate pentose ring impact on CFTR gating and hydrolysis. FEBS Lett 518: 183–188 - PubMed
-
- Aleksandrov L, Aleksandrov AA, Chang XB, Riordan JR 2002b. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J Biol Chem 277: 15419–15425 - PubMed
-
- Aleksandrov AA, Aleksandrov LA, Riordan JR 2007. CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflugers Arch 453: 693–702 - PubMed
-
- Aleksandrov L, Aleksandrov A, Riordan JR 2008. Mg2+-dependent ATP occlusion at the first nucleotide-binding domain (NBD1) of CFTR does not require the second (NBD2). Biochem J 416: 129–136 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical