

A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis
- PMID: 23380220
- PMCID: PMC4060894
- DOI: 10.1016/j.jaci.2012.11.053
A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis
Abstract
Background: Cigarette smoke-induced chronic obstructive pulmonary disease (COPD) is a life-threatening inflammatory disorder of the lung. The development of effective therapies for COPD has been hampered by the lack of an animal model that mimics the human disease in a short timeframe.
Objectives: We sought to create an early-onset mouse model of cigarette smoke-induced COPD that develops the hallmark features of the human condition in a short time-frame. We also sought to use this model to better understand pathogenesis and the roles of macrophages and mast cells (MCs) in patients with COPD.
Methods: Tightly controlled amounts of cigarette smoke were delivered to the airways of mice, and the development of the pathologic features of COPD was assessed. The roles of macrophages and MC tryptase in pathogenesis were evaluated by using depletion and in vitro studies and MC protease 6-deficient mice.
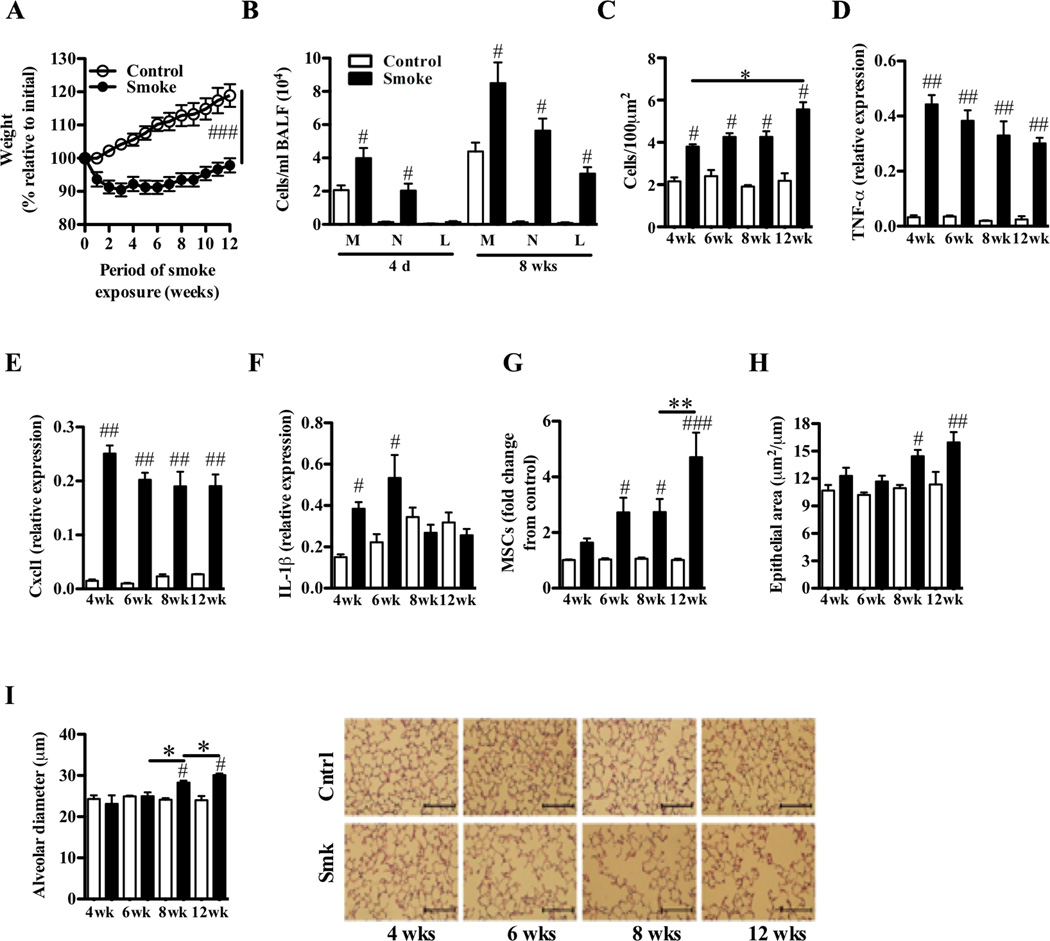
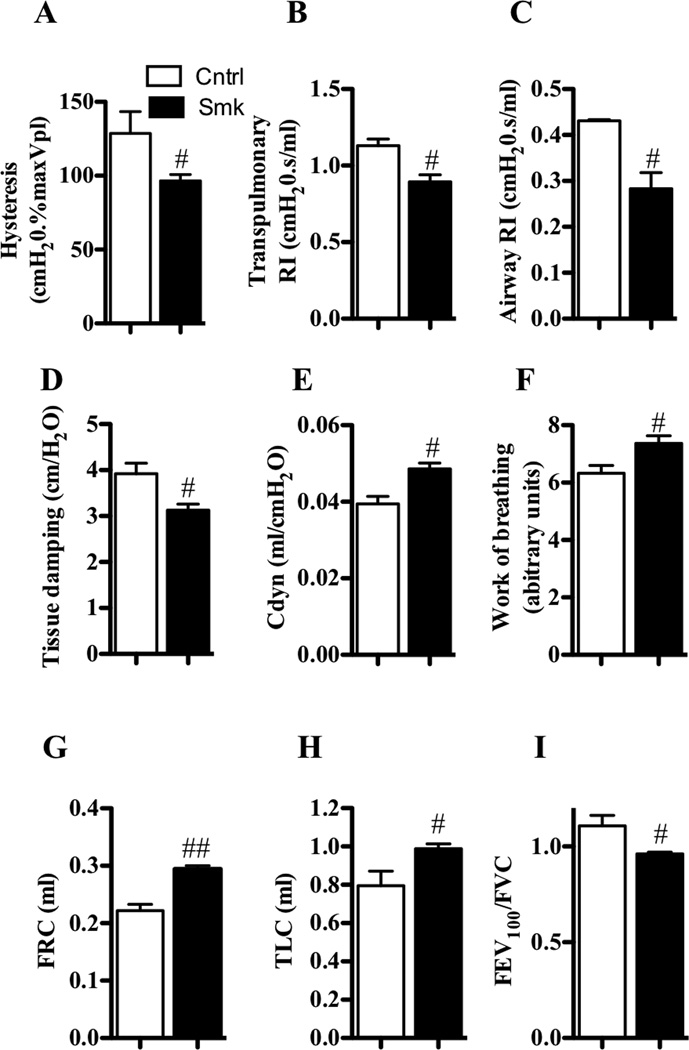
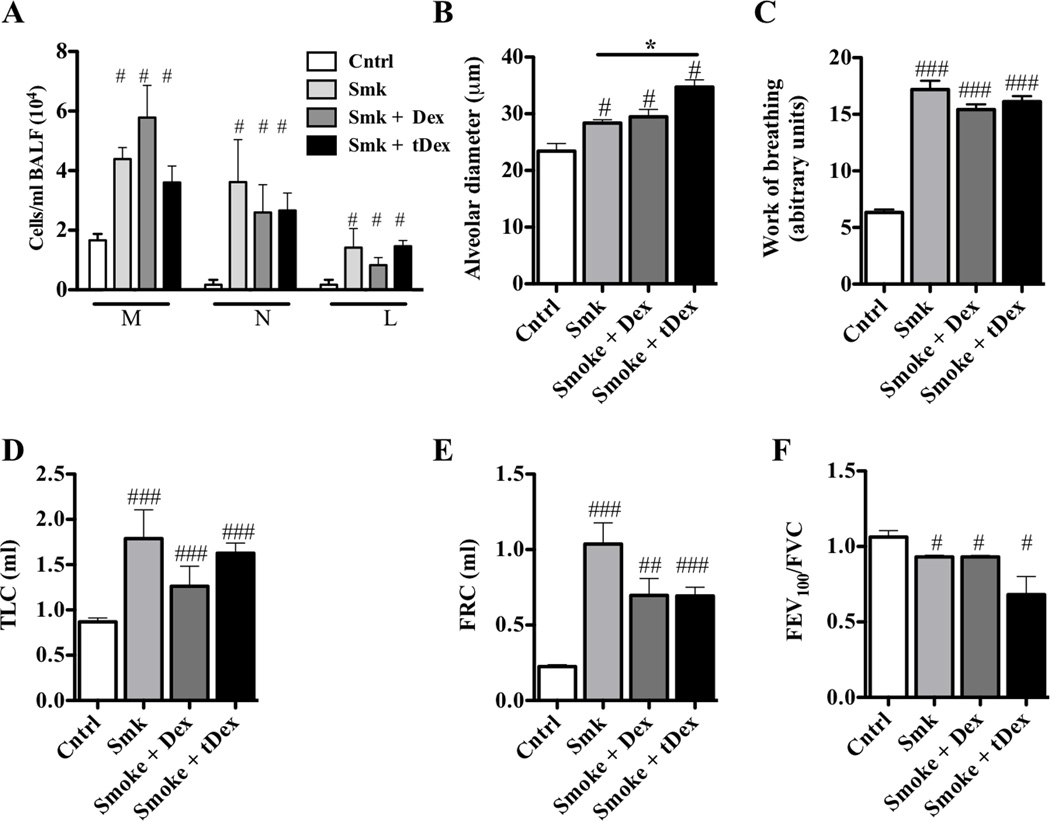
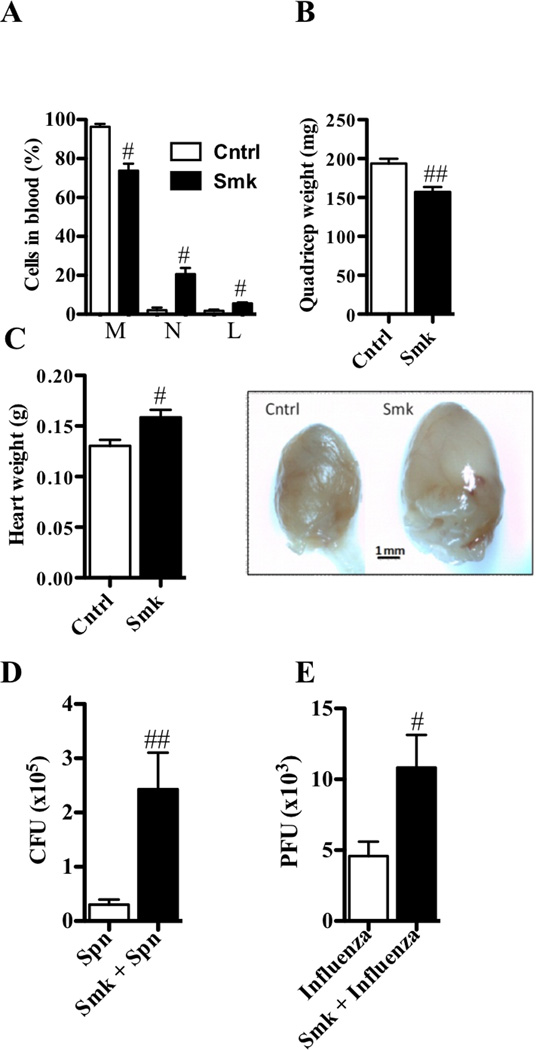
Results: After just 8 weeks of smoke exposure, wild-type mice had chronic inflammation, mucus hypersecretion, airway remodeling, emphysema, and reduced lung function. These characteristic features of COPD were glucocorticoid resistant and did not spontaneously resolve. Systemic effects on skeletal muscle and the heart and increased susceptibility to respiratory tract infections also were observed. Macrophages and tryptase-expressing MCs were required for the development of COPD. Recombinant MC tryptase induced proinflammatory responses from cultured macrophages.
Conclusion: A short-term mouse model of cigarette smoke-induced COPD was developed in which the characteristic features of the disease were induced more rapidly than in existing models. The model can be used to better understand COPD pathogenesis, and we show a requirement for macrophages and tryptase-expressing MCs.
Copyright © 2013 American Academy of Allergy, Asthma & Immunology. Published by Mosby, Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- WHO. Global Burden of Disease: 2004 Update. 2008
-
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. - PubMed
-
- Vlahos R, Bozinovski S, Gualano RC, Ernst M, Anderson GP. Modelling COPD in mice. Pulm Pharmacol Ther. 2006;19:12–17. - PubMed
-
- Bracke KR, D'hulst AI, Maes T, Moerloose KB, Demedts IK, Lebecque S, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol. 2006;177:4350–4359. - PubMed
-
- Gaschler GJ, Zavitz CCJ, Bauer CMT, Skrtic M, Lindahl M, Robbins CS, et al. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am J Respir Cell Mol Biol. 2007;38:218–226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
