

Signaling pathways of ESE-16, an antimitotic and anticarbonic anhydrase estradiol analog, in breast cancer cells
- PMID: 23382857
- PMCID: PMC3561402
- DOI: 10.1371/journal.pone.0053853
Signaling pathways of ESE-16, an antimitotic and anticarbonic anhydrase estradiol analog, in breast cancer cells
Erratum in
- PLoS One. 2013;8(10). doi: 10.1371/annotation/6afd5e63-46d6-42a1-b661-6688a04bfd16
Abstract
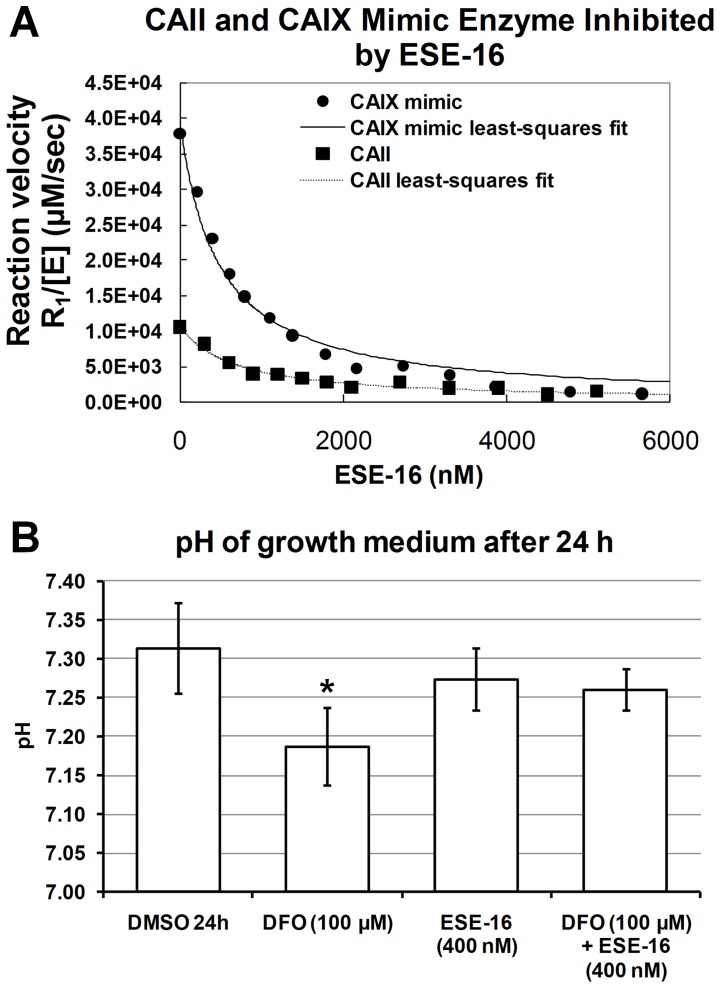
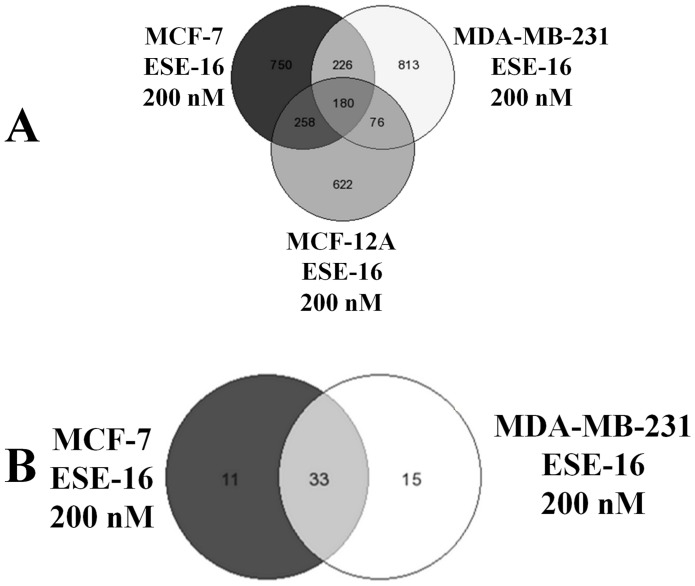
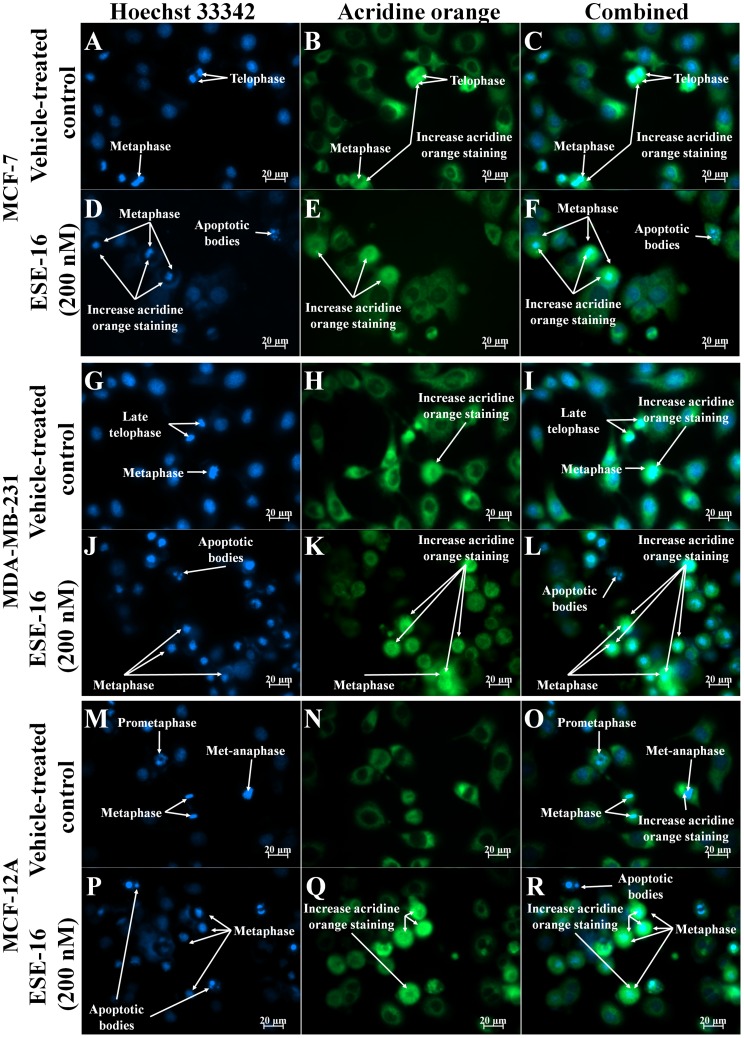
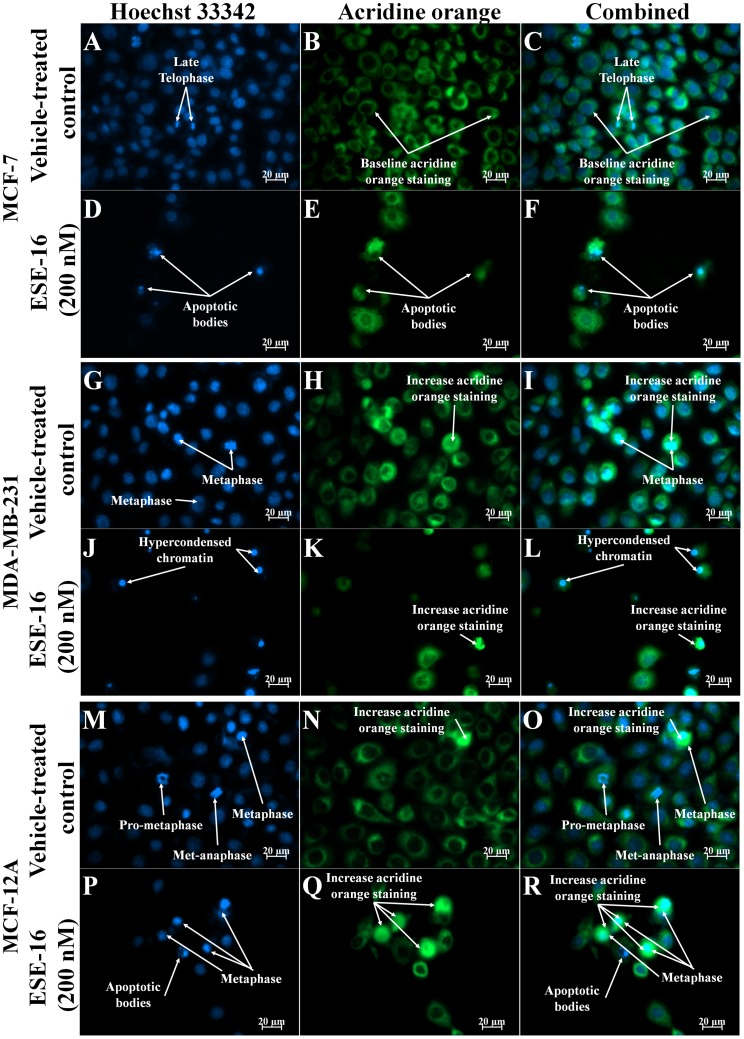
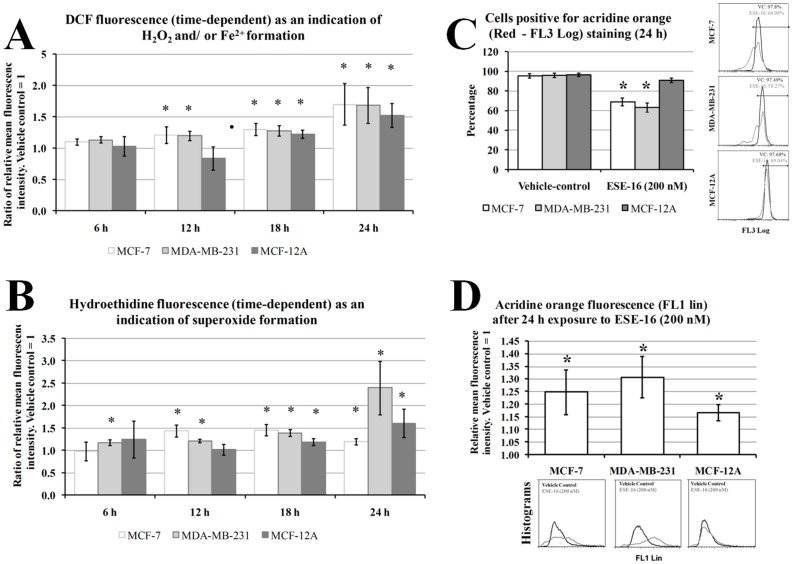
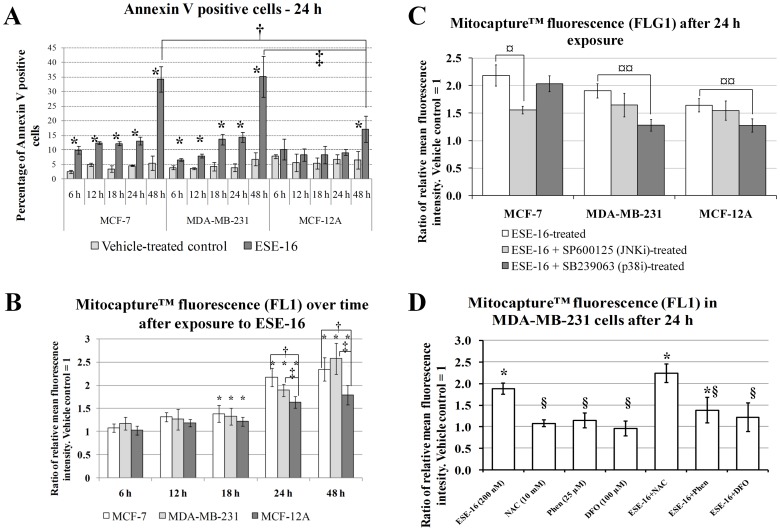
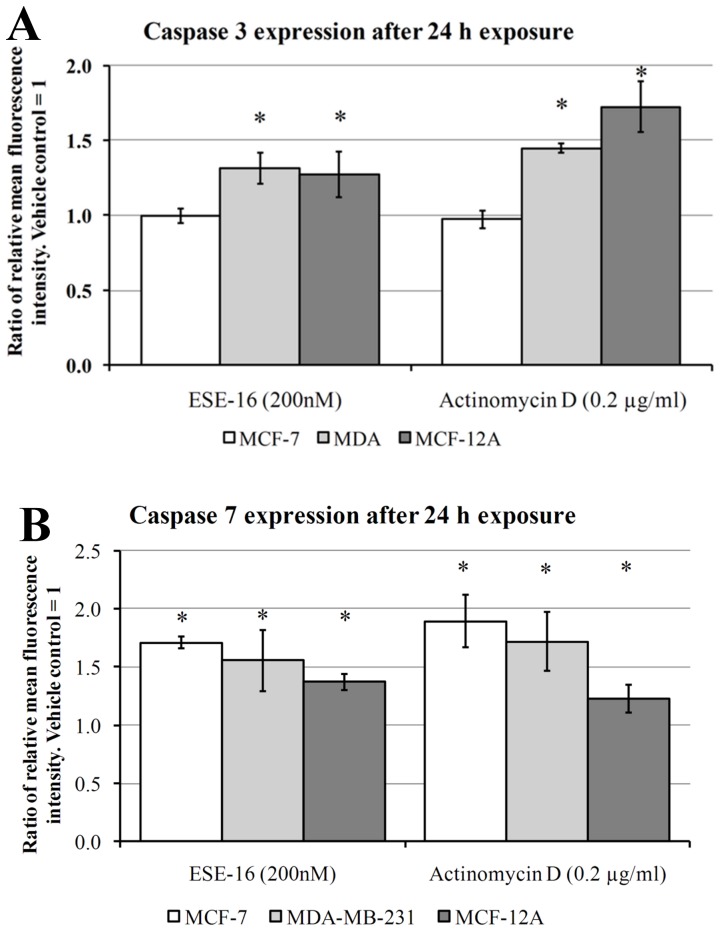
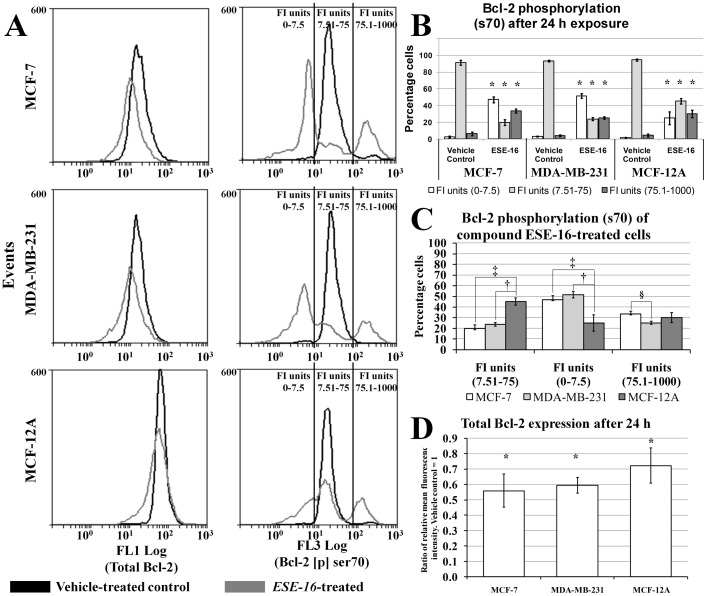
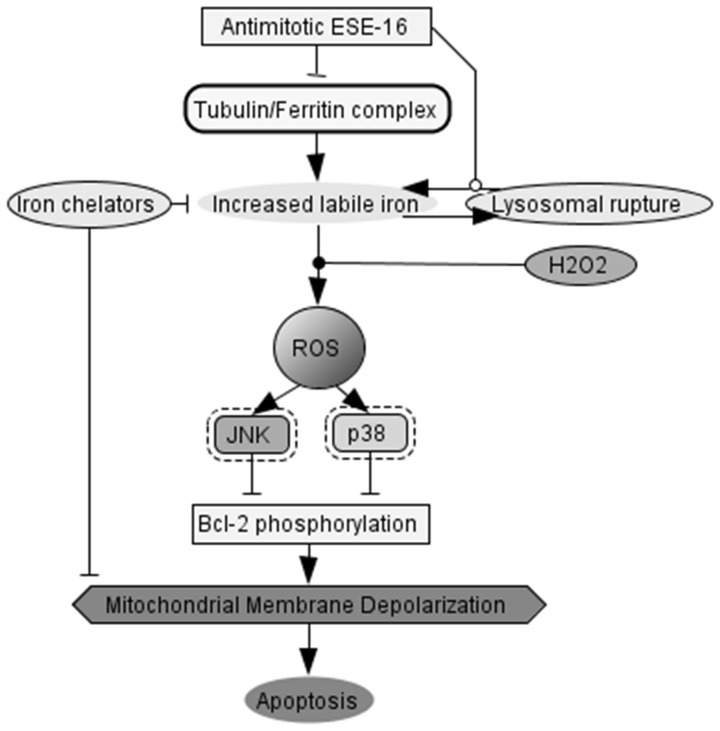
The aim of this study was to characterize the in vitro action of 2-ethyl-3-O-sulphamoyl-estra-1,3,5(10)16-tetraene (ESE-16) on non-tumorigenic MCF-12A, tumorigenic MCF-7 and metastatic MDA-MB-231 breast cancer cells. ESE-16 is able to inhibit the activity of a carbonic anhydrase II and a mimic of carbonic anhydrase IX in the nanomolar range. Gene and protein expression studies using various techniques including gene and antibody microarrays and various flow cytometry assays yielded valuable information about the mechanism of action of ESE-16. The JNK pathway was identified as an important pathway mediating the effects of ESE-16 while the p38 stress-induced pathway is more important in MDA-MB-231 cells exposed to ESE-16. Lysosomal rupture and iron metabolism was identified as important mediators of mitochondrial membrane depolarization. Abrogation of Bcl-2 phosphorylation status as a result of ESE-16 also plays a role in inducing mitochondrial membrane depolarization. The study provides a basis for future research projects to develop the newly synthesized compound into a clinically usable anticancer agent either alone or in combination with other agents.
Keywords: Antimitotic, anticarbonic anhydrase IX, apoptosis, autophagy, cell cycle arrest, Bcl-2, JNK, p38, mitochondrial membrane depolarization, flow cytometry, gene expression and protein microarray, anticancer.
Conflict of interest statement
Figures
References
-
- Bughani U, Li S, Joshi HC (2009) Recent patents reveal microtubules as persistent promising target for novel drug development for cancers. Recent Pat Antiinfect Drug Discov 4: 164–182. - PubMed
-
- Newman SP, Ireson CR, Tutill HJ, Day JM, Parsons MF, et al. (2006) The role of 17beta-hydroxysteroid dehydrogenases in modulating the activity of 2-methoxyestradiol in breast cancer cells. Cancer Res 66: 324–330. - PubMed
-
- Supuran CT, Scozzafava A (2007) Carbonic anhydrases as targets for medicinal chemistry. Bioorganic & Medicinal Chemistry 15: 4336–4350. - PubMed
-
- Snoeks TJ, Mol IM, Que I, Kaijzel EL, Lowik CW (2011) 2-methoxyestradiol analogue ENMD-1198 reduces breast cancer-induced osteolysis and tumor burden both in vitro and in vivo. Molecular Cancer Therapeutics 10: 874–882. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
