

De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome
- PMID: 23383720
- PMCID: PMC3707024
- DOI: 10.1186/gm415
De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome
Abstract
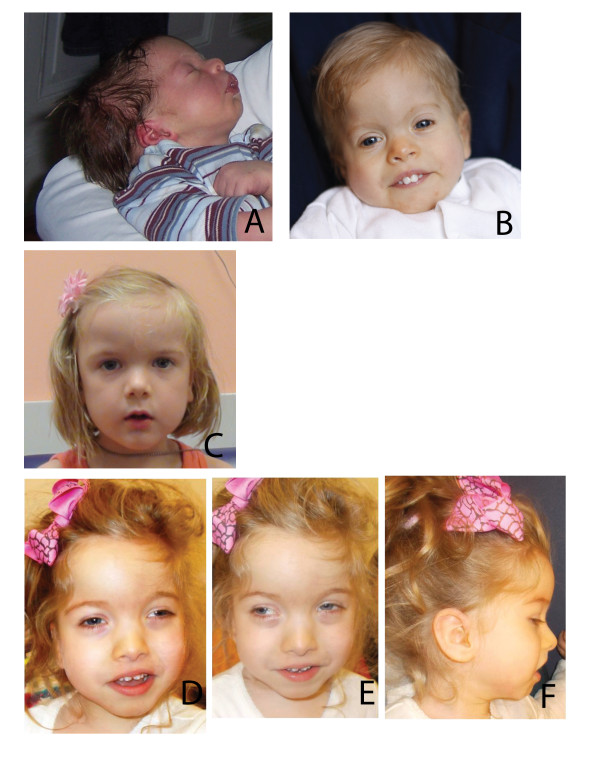
Background: Molecular diagnostics can resolve locus heterogeneity underlying clinical phenotypes that may otherwise be co-assigned as a specific syndrome based on shared clinical features, and can associate phenotypically diverse diseases to a single locus through allelic affinity. Here we describe an apparently novel syndrome, likely caused by de novo truncating mutations in ASXL3, which shares characteristics with Bohring-Opitz syndrome, a disease associated with de novo truncating mutations in ASXL1.
Methods: We used whole-genome and whole-exome sequencing to interrogate the genomes of four subjects with an undiagnosed syndrome.
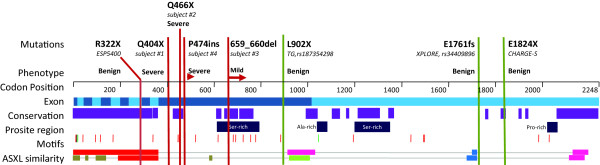
Results: Using genome-wide sequencing, we identified heterozygous, de novo truncating mutations in ASXL3, a transcriptional repressor related to ASXL1, in four unrelated probands. We found that these probands shared similar phenotypes, including severe feeding difficulties, failure to thrive, and neurologic abnormalities with significant developmental delay. Further, they showed less phenotypic overlap with patients who had de novo truncating mutations in ASXL1.
Conclusion: We have identified truncating mutations in ASXL3 as the likely cause of a novel syndrome with phenotypic overlap with Bohring-Opitz syndrome.
Figures
References
-
- Hastings R, Cobben JM, Gillessen-Kaesbach G, Goodship J, Hove H, Kjaergaard S, Kemp H, Kingston H, Lunt P, Mansour S, McGowan R, Metcalfe K, Murdoch-Davis C, Ray M, Rio M, Smithson S, Tolmie J, Turnpenny P, van Bon B, Wieczorek D, Newbury-Ecob R. Bohring-Opitz (Oberklaid-Danks) syndrome: clinical study, review of the literature, and discussion of possible pathogenesis. Eur J Hum Genet. 2011;19:513–519. doi: 10.1038/ejhg.2010.234. - DOI - PMC - PubMed
-
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, de Vries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R, Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BB. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat GenetNature genetics. 2011;43:729–731. doi: 10.1038/ng.868. - DOI - PubMed
-
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, Dahl F, Fernandez A, Staker B, Pant KP, Baccash J, Borcherding AP, Brownley A, Cedeno R, Chen L, Chernikoff D, Cheung A, Chirita R, Curson B, Ebert JC, Hacker CR, Hartlage R, Hauser B, Huang S, Jiang Y, Karpinchyk V. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science (New York, NY) 2010;327:78–81. doi: 10.1126/science.1181498. - DOI - PubMed
-
- Carnevali P, Baccash J, Halpern AL, Nazarenko I, Nilsen GB, Pant KP, Ebert JC, Brownley A, Morenzoni M, Karpinchyk V, Martin B, Ballinger DG, Drmanac R. Computational techniques for human genome resequencing using mated gapped reads. J Comput Biol. 2012;19:279–292. doi: 10.1089/cmb.2011.0201. - DOI - PubMed
-
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Puttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weissmann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi SG, Cohen M, Fattahi Z. et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
