

Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review
- PMID: 23384594
- PMCID: PMC3647006
- DOI: 10.1016/j.pharmthera.2013.01.016
Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review
Abstract
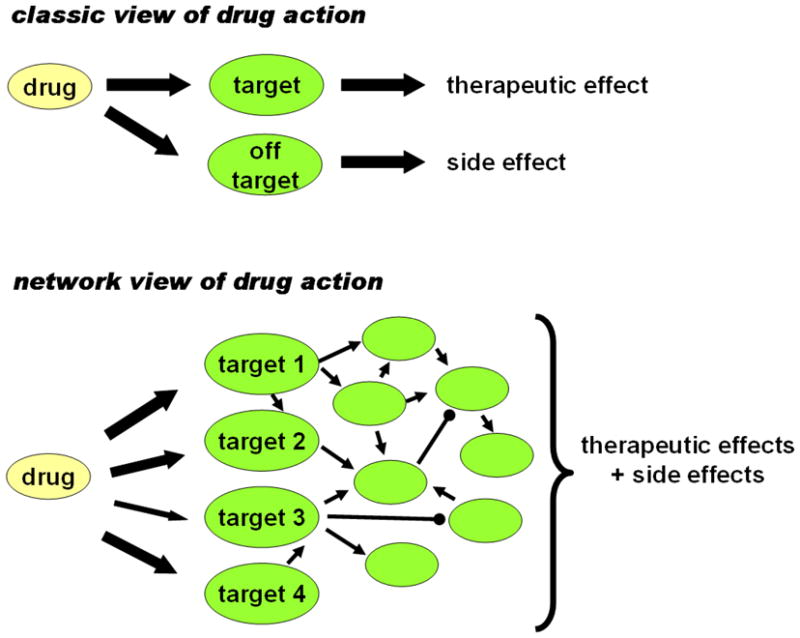
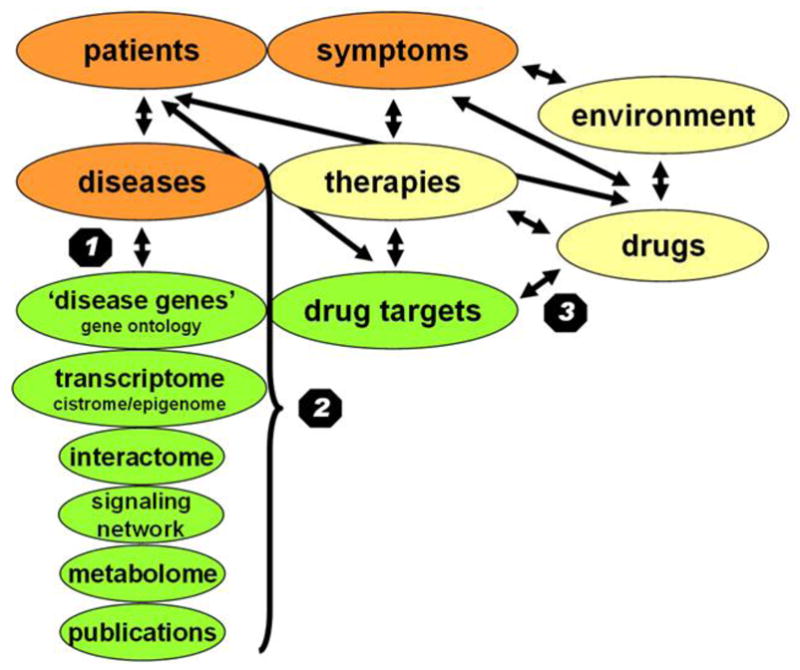
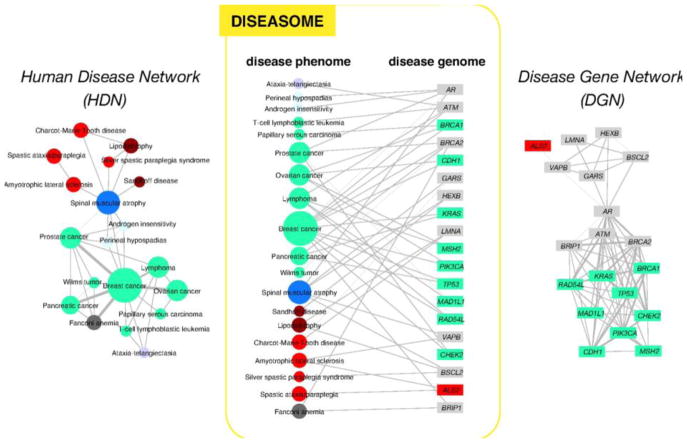
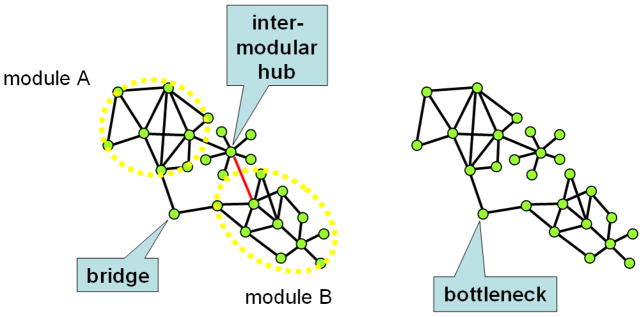
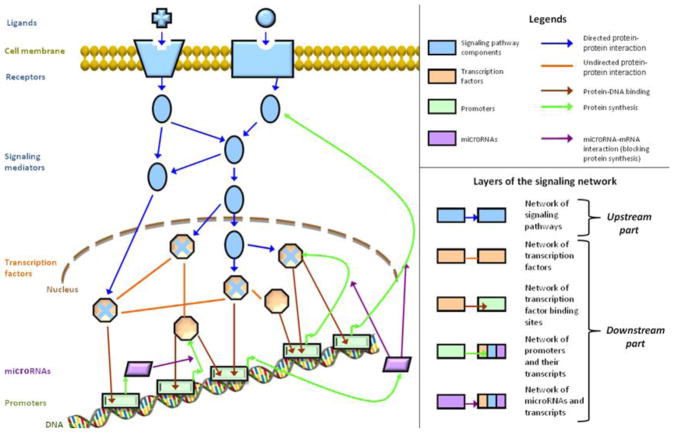
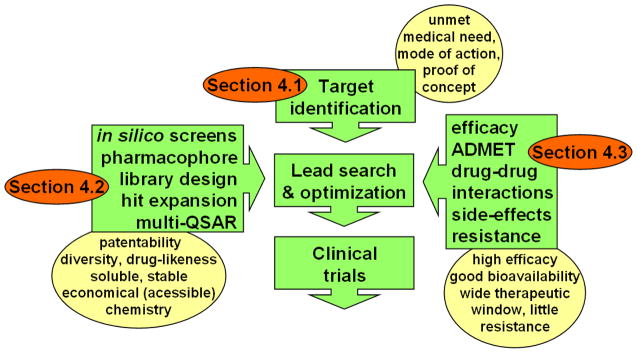
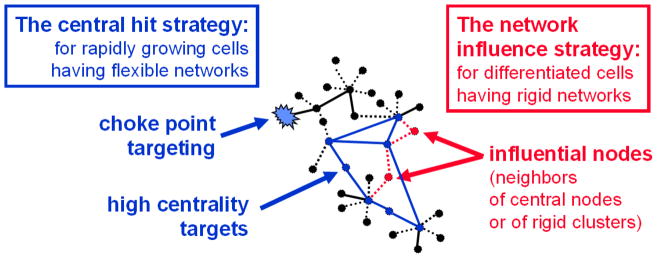
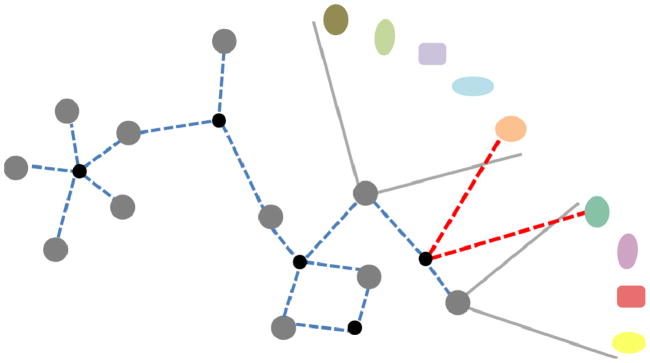
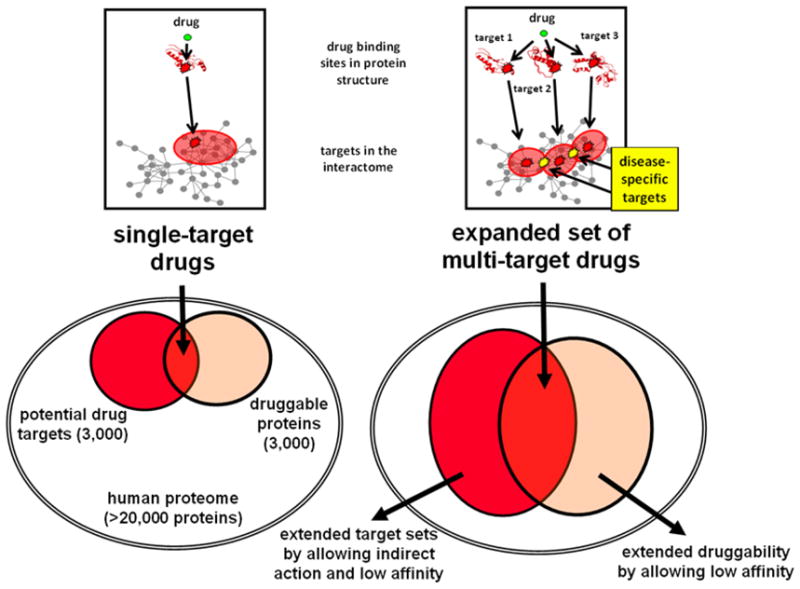
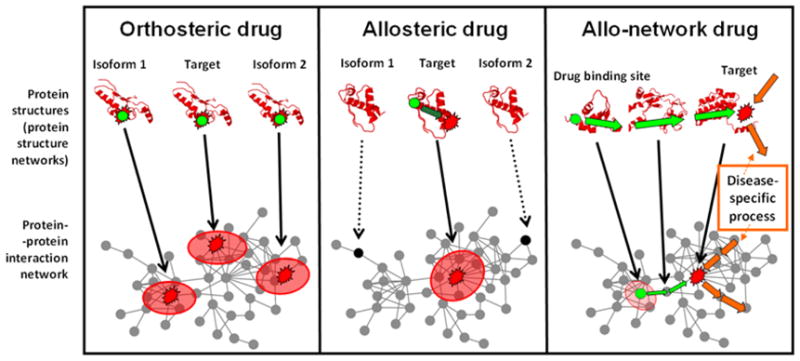
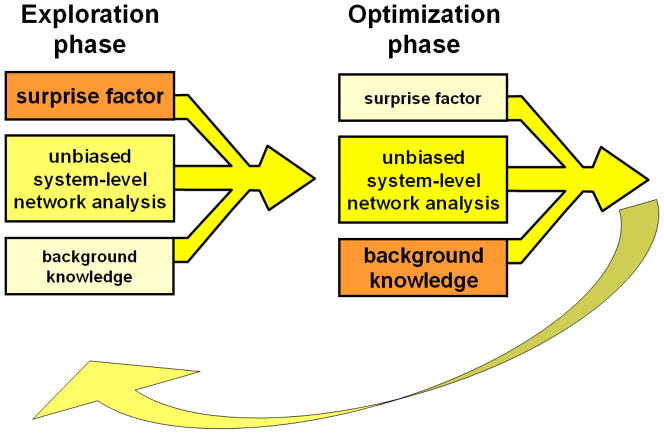
Despite considerable progress in genome- and proteome-based high-throughput screening methods and in rational drug design, the increase in approved drugs in the past decade did not match the increase of drug development costs. Network description and analysis not only give a systems-level understanding of drug action and disease complexity, but can also help to improve the efficiency of drug design. We give a comprehensive assessment of the analytical tools of network topology and dynamics. The state-of-the-art use of chemical similarity, protein structure, protein-protein interaction, signaling, genetic interaction and metabolic networks in the discovery of drug targets is summarized. We propose that network targeting follows two basic strategies. The "central hit strategy" selectively targets central nodes/edges of the flexible networks of infectious agents or cancer cells to kill them. The "network influence strategy" works against other diseases, where an efficient reconfiguration of rigid networks needs to be achieved by targeting the neighbors of central nodes/edges. It is shown how network techniques can help in the identification of single-target, edgetic, multi-target and allo-network drug target candidates. We review the recent boom in network methods helping hit identification, lead selection optimizing drug efficacy, as well as minimizing side-effects and drug toxicity. Successful network-based drug development strategies are shown through the examples of infections, cancer, metabolic diseases, neurodegenerative diseases and aging. Summarizing >1200 references we suggest an optimized protocol of network-aided drug development, and provide a list of systems-level hallmarks of drug quality. Finally, we highlight network-related drug development trends helping to achieve these hallmarks by a cohesive, global approach.
Copyright © 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
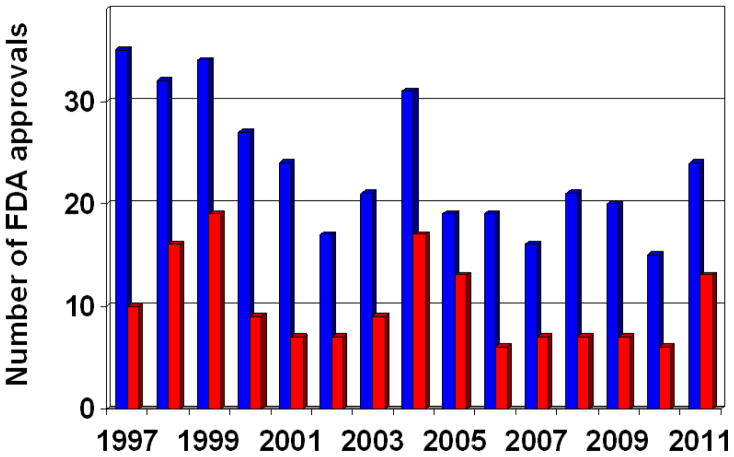
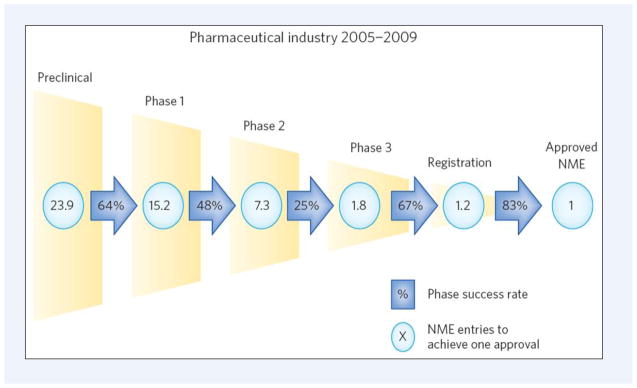
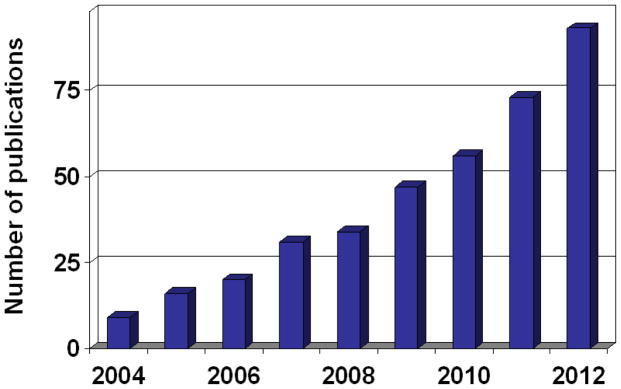
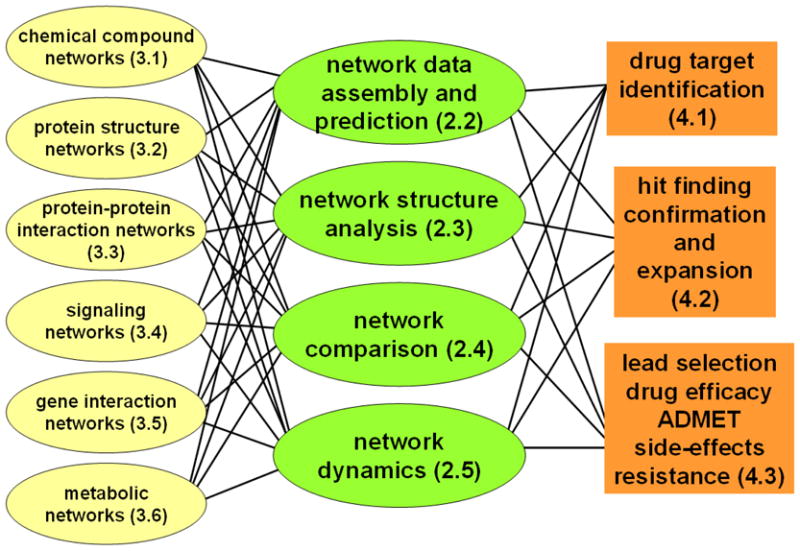
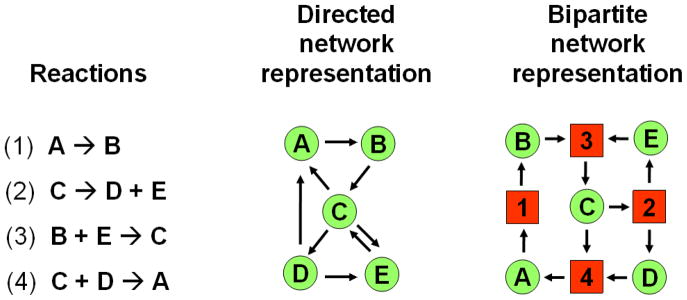
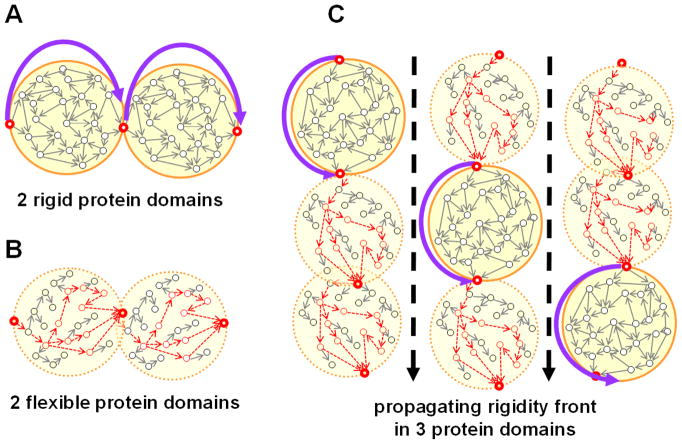
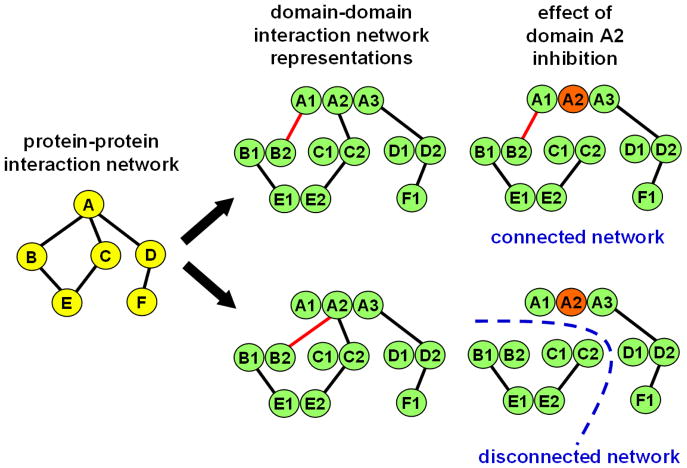
References
-
- Abdi A, Tahoori MB, Emamian ES. Fault diagnosis engineering of digital circuits can identify vulnerable molecules in complex cellular pathways. Science Signaling. 2008;1:ra10. - PubMed
-
- Adamcsek B, Palla G, Farkas IJ, Derenyi I, Vicsek T. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics. 2006;22:1021–1023. - PubMed
-
- Adar E. GUESS: A language and interface for graph exploration. In: Grinter R, Rodden T, Aoki P, Cutrell E, Jeffries R, Olson G, editors. CHI ‘06 Proceedings of the SIGCHI conference on human factors in computing systems. New York: Association for Computing Machinery; 2006. pp. 791–800.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
