

Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders
- PMID: 23386033
- PMCID: PMC3778343
- DOI: 10.1038/ejhg.2012.305
Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders
Abstract
Joubert syndrome and related disorders (JSRD) are clinically and genetically heterogeneous ciliopathies sharing a peculiar midbrain-hindbrain malformation known as the 'molar tooth sign'. To date, 19 causative genes have been identified, all coding for proteins of the primary cilium. There is clinical and genetic overlap with other ciliopathies, in particular with Meckel syndrome (MKS), that is allelic to JSRD at nine distinct loci. We previously identified the INPP5E gene as causative of JSRD in seven families linked to the JBTS1 locus, yet the phenotypic spectrum and prevalence of INPP5E mutations in JSRD and MKS remain largely unknown. To address this issue, we performed INPP5E mutation analysis in 483 probands, including 408 JSRD patients representative of all clinical subgroups and 75 MKS fetuses. We identified 12 different mutations in 17 probands from 11 JSRD families, with an overall 2.7% mutation frequency among JSRD. The most common clinical presentation among mutated families (7/11, 64%) was Joubert syndrome with ocular involvement (either progressive retinopathy and/or colobomas), while the remaining cases had pure JS. Kidney, liver and skeletal involvement were not observed. None of the MKS fetuses carried INPP5E mutations, indicating that the two ciliopathies are not allelic at this locus.
Figures
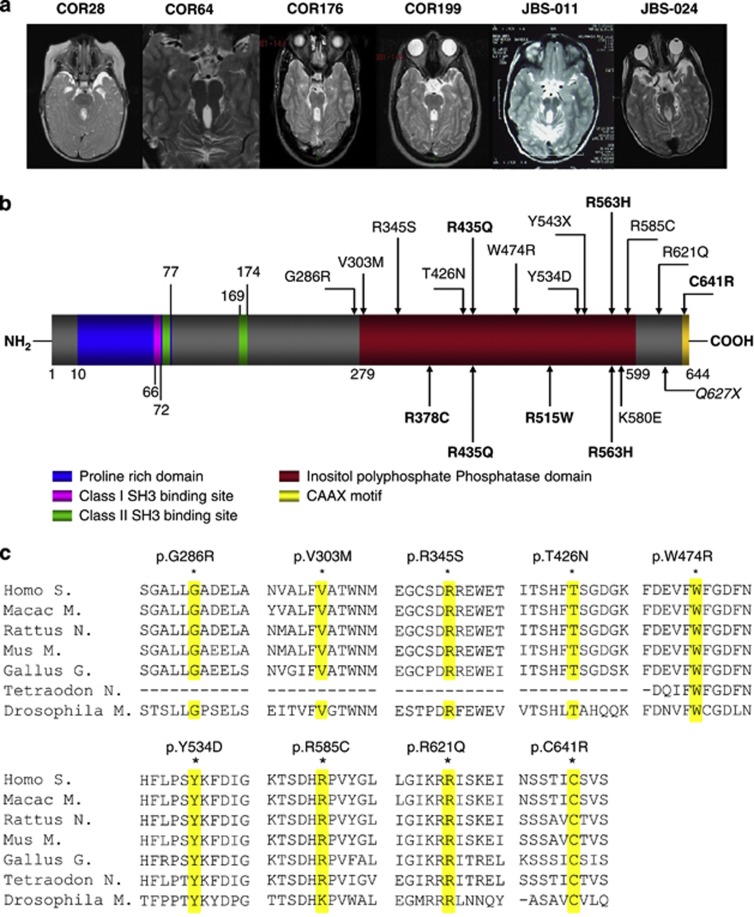
References
-
- Parisi M, Glass I.Joubert Syndrome and Related Disorders: GeneReviews http://www.ncbi.nlm.nih.gov/books/NBK1325/ Initial Posting: 9 July 2003; Last Revision: 14 June 2012.
-
- Srour M, Hamdan FF, Schwartzentruber JA, et al. Mutation in TMEM231 cause Joubert Syndrome in French Canadians. J Med Genet. 2012;49:636–641. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
