

Novel allelic variants and evidence for a prevalent mutation in URAT1 causing renal hypouricemia: biochemical, genetics and functional analysis
- PMID: 23386035
- PMCID: PMC3778361
- DOI: 10.1038/ejhg.2013.3
Novel allelic variants and evidence for a prevalent mutation in URAT1 causing renal hypouricemia: biochemical, genetics and functional analysis
Abstract
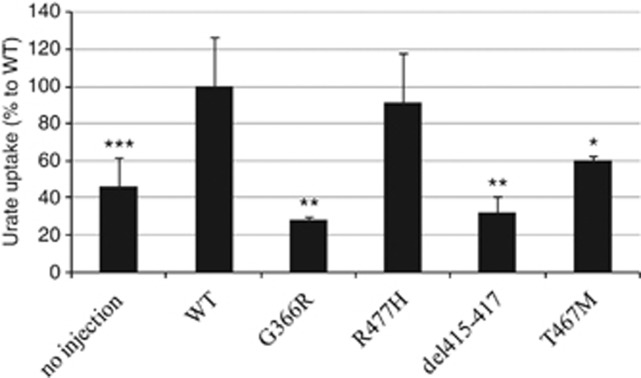
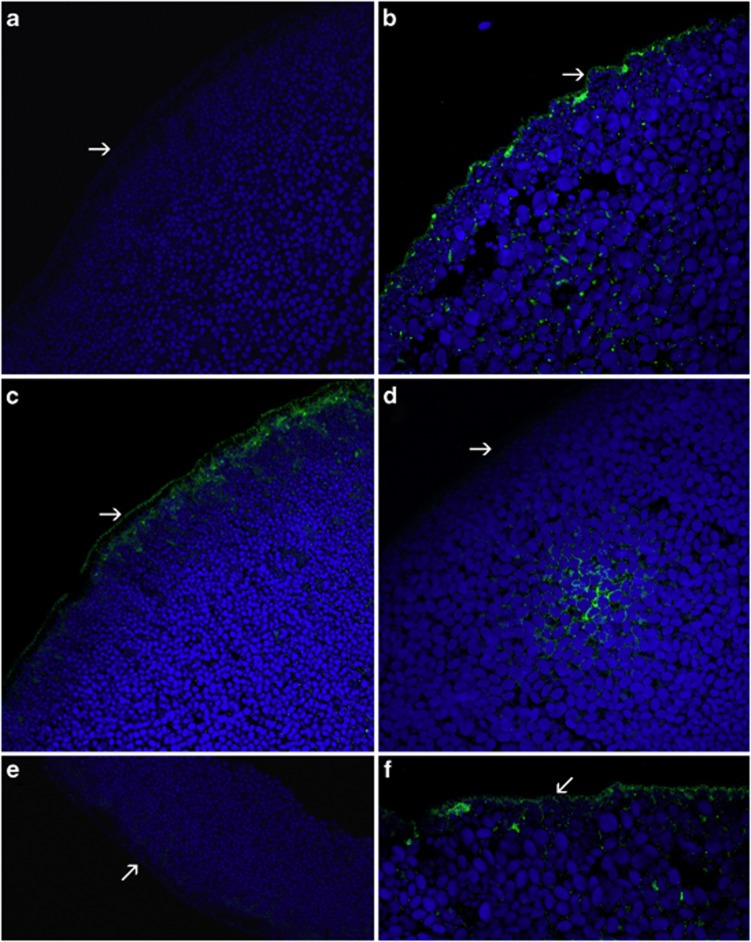
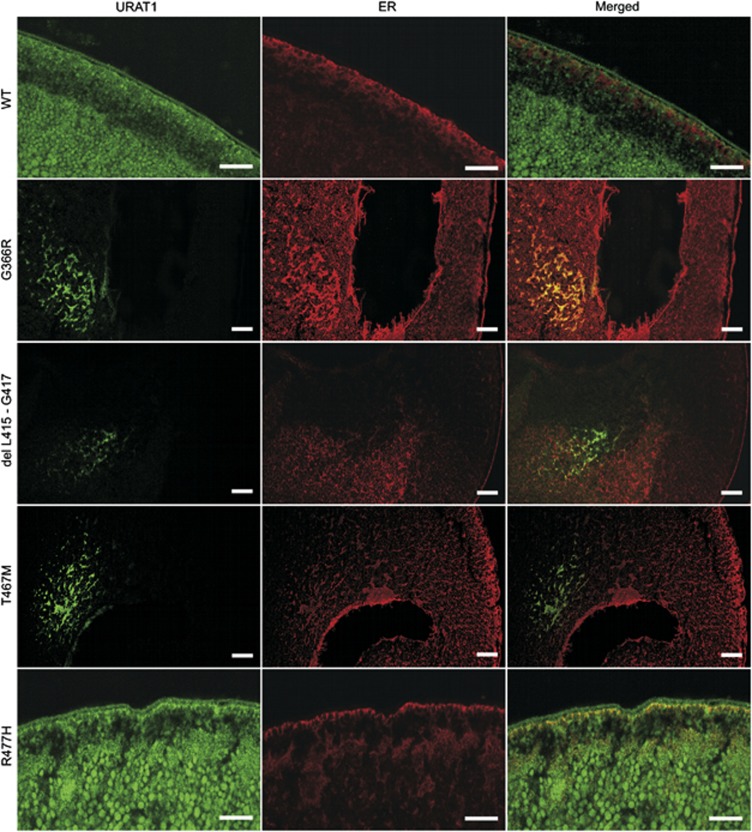
Renal hypouricemia (RHUC) is a heterogeneous inherited disorder characterized by impaired tubular uric acid (UA) transport with severe complications, such as acute kidney injury (AKI). Type 1 is caused by a loss-of-function mutation in the SLC22A12 gene (URAT1), type 2 in the SLC2A9 gene (GLUT9). This article describes three Czech families with RHUC type 1. The serum UA in the probands was 0.9, 1.1 and 0.5 mg/dl and expressed as an increase in the fractional excretion of UA (48, 43 and 39%). The sequencing analysis of SLC22A12 revealed three novel variants: p.G366R, p.T467M and a deletion p.L415_G417del. A detailed metabolic investigation in proband C for progressive visual failure supported suspicion of neuronal ceroid lipofuscinosis type 7 conditioned by the mutation in the MFSD8 gene. Functional studies showed significantly decreased urate uptake and a mis-localized URAT1 signal in p.G366R, p.L415_G417del and p.T467M. Furthermore, colocalization studies showed accumulation of URAT1 protein in the endoplasmic reticulum. The findings suggest that loss-of-function mutations cause RHUC via loss of UA absorption partly by protein misfolding. However, they do not necessarily lead to AKI and a possible genotype-phenotype correlation was not proposed. Furthermore, results confirm an uneven geographical and ethnic distribution of SLC22A12 variants; the p.L415_G417del mutation predominates in the Roma ethnic group in the Czech Republic.
Figures

References
-
- Watanabe S, Kang DH, Feng L, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension. 2002;40:355–360. - PubMed
-
- Sotgiu S, Pugliatti M, Sanna A, et al. Serum uric acid and multiple sclerosis. Neurol Sci. 2002;23:183–188. - PubMed
-
- Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. - PubMed
-
- Augustin R, Carayannopoulos MO, Dowd L, Phay JE, Moley JF, Moley KH. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J Biol Chem. 2004;279:16229–16236. - PubMed
-
- Ichida K, Hosoyamada M, Hisatome I, et al. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol. 2004;15:164–173. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
