

Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation
- PMID: 23386971
- PMCID: PMC3564041
- DOI: 10.1038/srep01215
Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation
Abstract
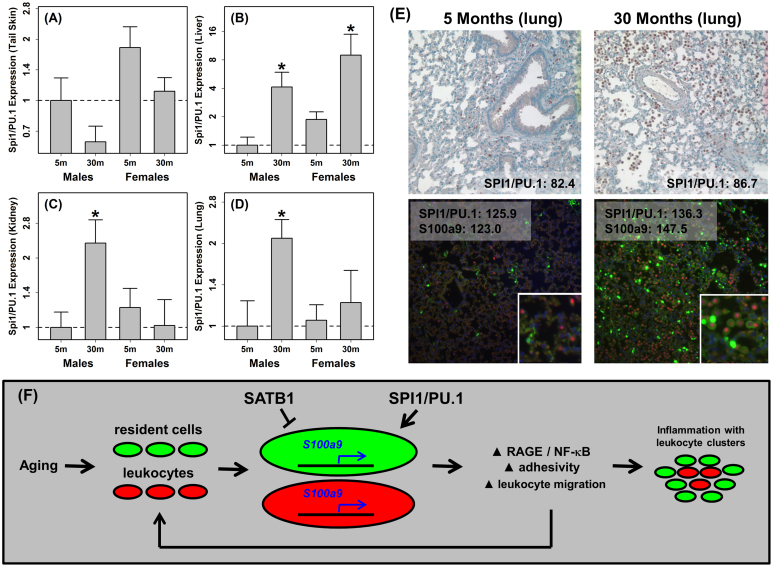
The S100a8 and S100a9 genes encode a pro-inflammatory protein (calgranulin) that has been implicated in multiple diseases. However, involvement of S100a8/a9 in the basic mechanisms of intrinsic aging has not been established. In this study, we show that shifts in the abundance of S100a8 and S100a9 mRNA are a robust feature of aging in mammalian tissues, involving a range of cell types including the central nervous system. To identify transcription factors that control S100a9 expression, we performed a large-scale transcriptome analysis of 62 mouse and human cell types. We identified cell type-specific trends, as well as robust associations linking S100a9 coexpression to elevated frequency of ETS family motifs, and in particular, to motifs recognized by the transcription factor SPI/PU.1. Sparse occurrence of SATB1 motifs was also a strong predictor of S100a9 coexpression. These findings offer support for a novel mechanism by which a SPI1/PU.1-S100a9 axis sustains chronic inflammation during aging.
Figures
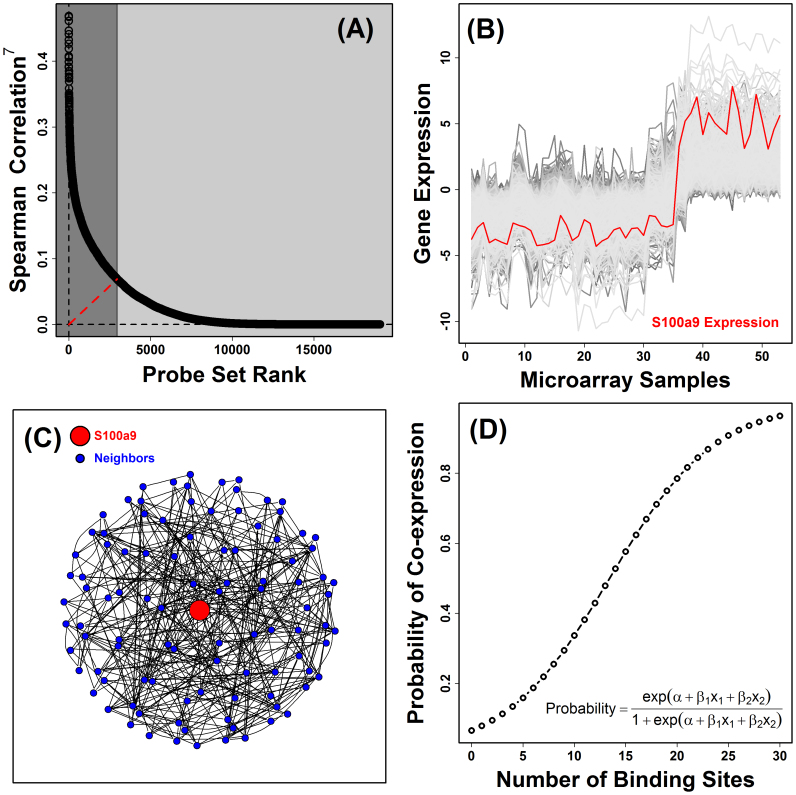
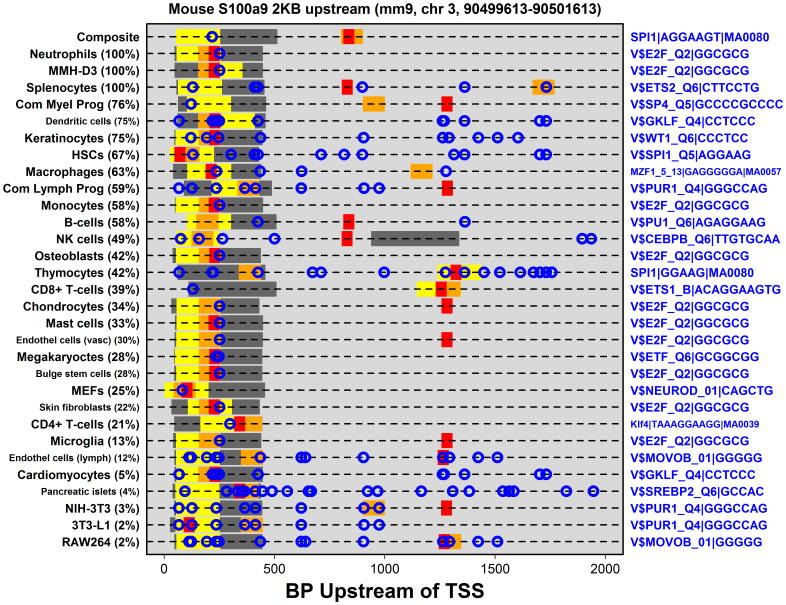
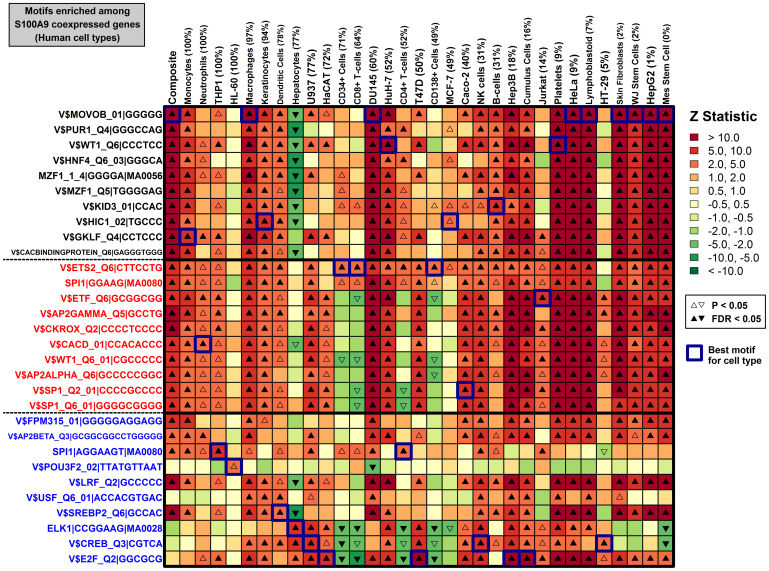
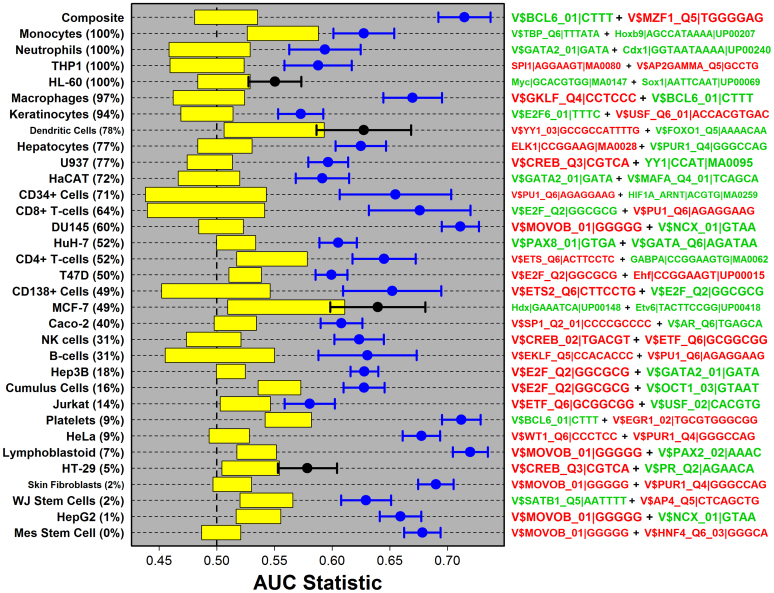
 . The dashed red line shown in (A) represents the segment with minimal distance between the origin (lower left corner) and the curve shown in the figure. This red line serves to define the foreground set of S100a9-coexpressed genes (dark grey region). In part (B), this S100a9-coexpression cluster is illustrated with respect to the 53 microarray samples used to calculate Spearman rank correlations shown in (A), where each microarray sample was generated by hybridization with cDNA derived from mouse chondrocytes. The foreground set of S100a9-coexpressed genes can thus be viewed as the local sub-network that surrounds S100a9, as illustrated in (C). In the final step (D), a generalized additive logistic model (GAM) is used identify significant associations between S100a9 coexpression and the number of TF binding sites present within the 2 KB region upstream of the transcription start site (or other genomic regions). In GAM models, the probability of S100a9 coexpression is modeled (vertical axis) as a function of two variables x1 and x2, where x1 is the length of unmasked sequence scanned for a given gene and x2 is the number of TF binding sites identified in the upstream region. GAM models were fit for each of 1209 TF binding sites, and a significant association between S100a9 coexpression and binding site occurrence was evaluated based upon significance of the coefficient β2.
. The dashed red line shown in (A) represents the segment with minimal distance between the origin (lower left corner) and the curve shown in the figure. This red line serves to define the foreground set of S100a9-coexpressed genes (dark grey region). In part (B), this S100a9-coexpression cluster is illustrated with respect to the 53 microarray samples used to calculate Spearman rank correlations shown in (A), where each microarray sample was generated by hybridization with cDNA derived from mouse chondrocytes. The foreground set of S100a9-coexpressed genes can thus be viewed as the local sub-network that surrounds S100a9, as illustrated in (C). In the final step (D), a generalized additive logistic model (GAM) is used identify significant associations between S100a9 coexpression and the number of TF binding sites present within the 2 KB region upstream of the transcription start site (or other genomic regions). In GAM models, the probability of S100a9 coexpression is modeled (vertical axis) as a function of two variables x1 and x2, where x1 is the length of unmasked sequence scanned for a given gene and x2 is the number of TF binding sites identified in the upstream region. GAM models were fit for each of 1209 TF binding sites, and a significant association between S100a9 coexpression and binding site occurrence was evaluated based upon significance of the coefficient β2.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
