

Calmodulin mutations associated with recurrent cardiac arrest in infants
- PMID: 23388215
- PMCID: PMC3834768
- DOI: 10.1161/CIRCULATIONAHA.112.001216
Calmodulin mutations associated with recurrent cardiac arrest in infants
Abstract
Background: Life-threatening disorders of heart rhythm may arise during infancy and can result in the sudden and tragic death of a child. We performed exome sequencing on 2 unrelated infants presenting with recurrent cardiac arrest to discover a genetic cause.
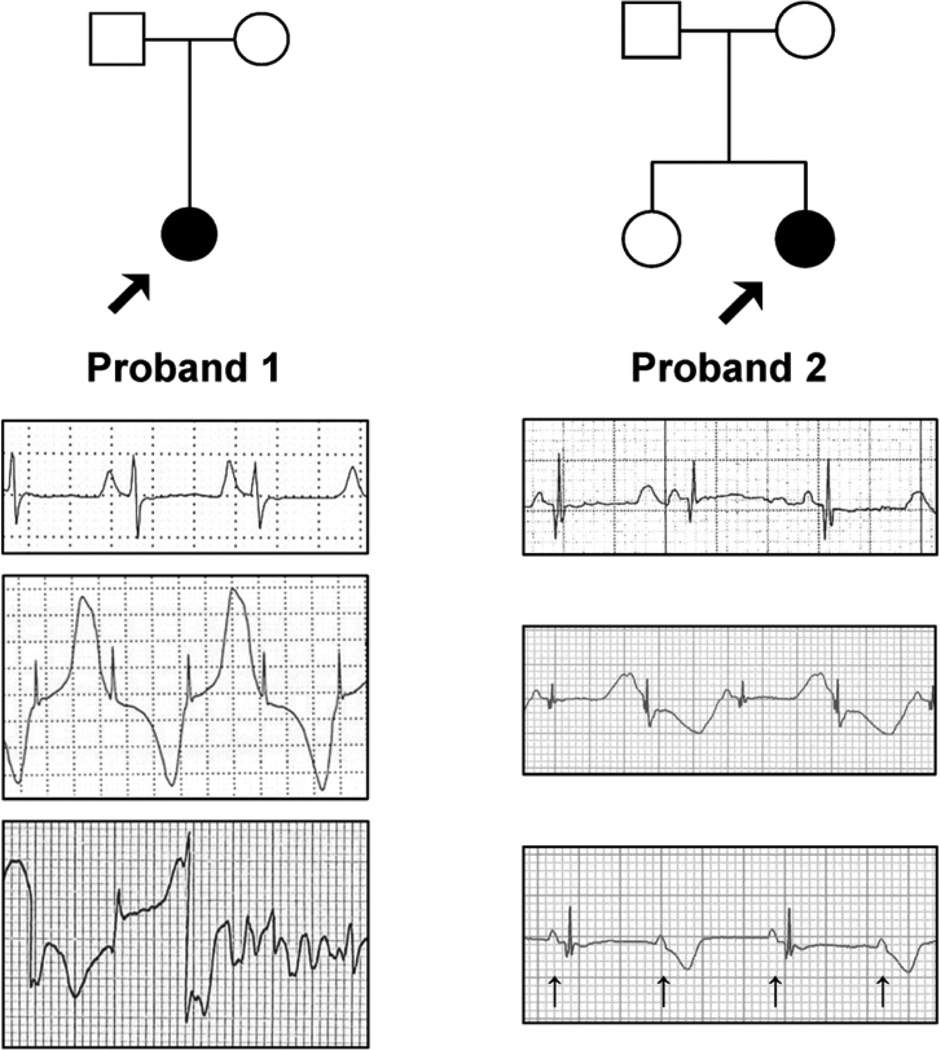
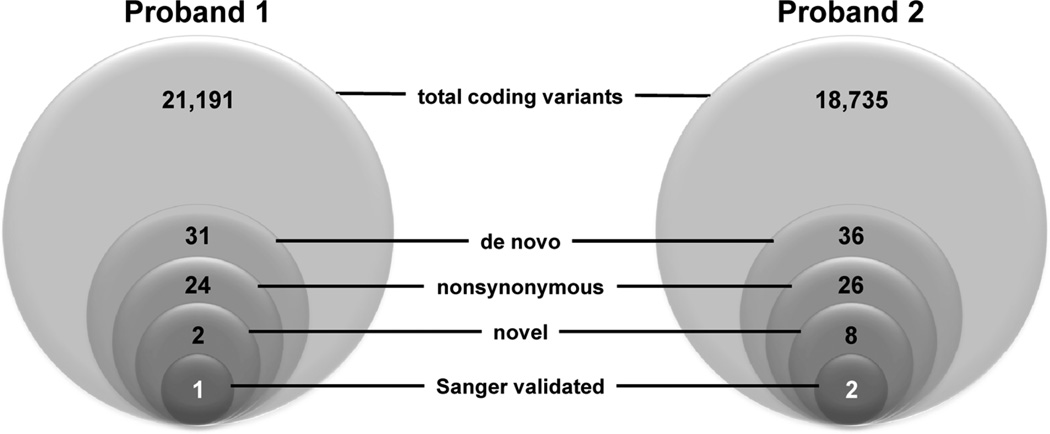
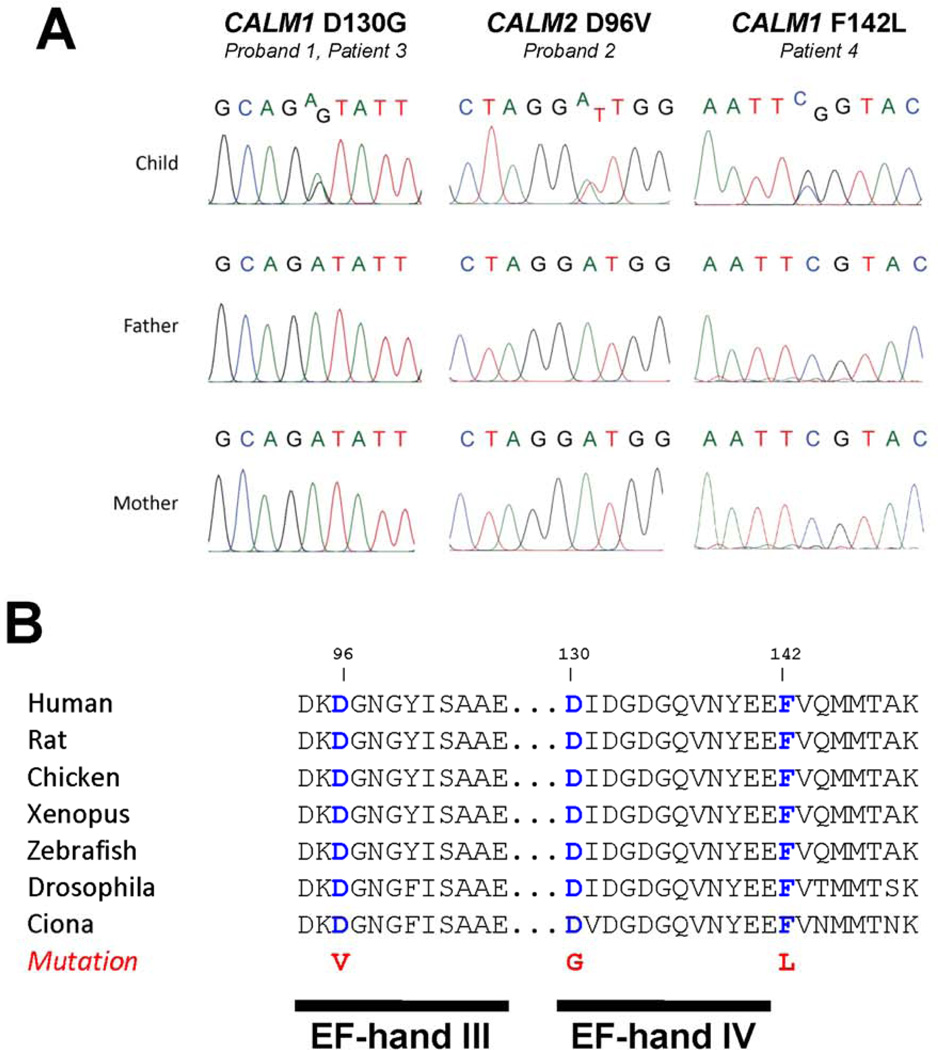
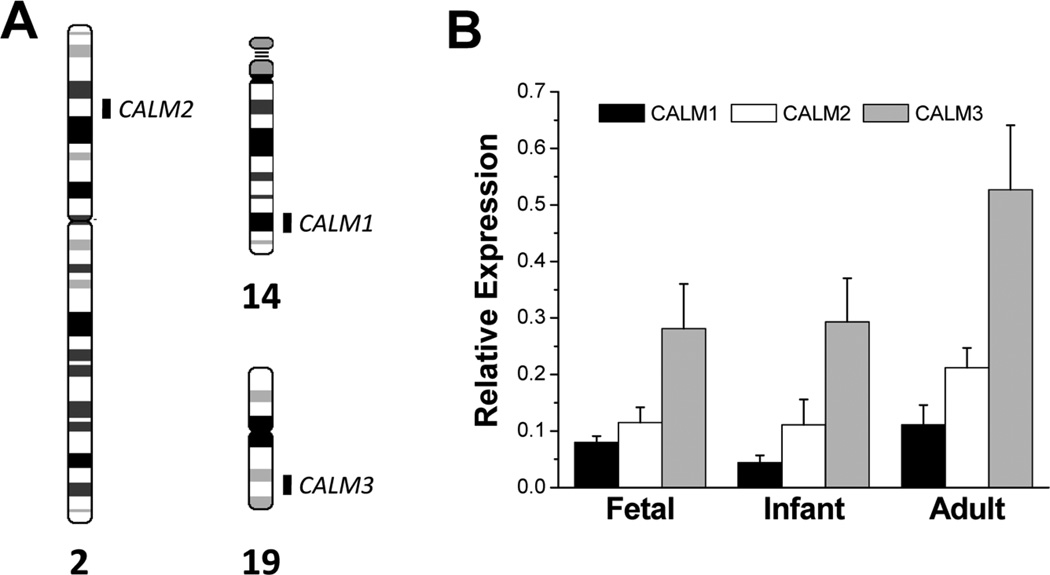
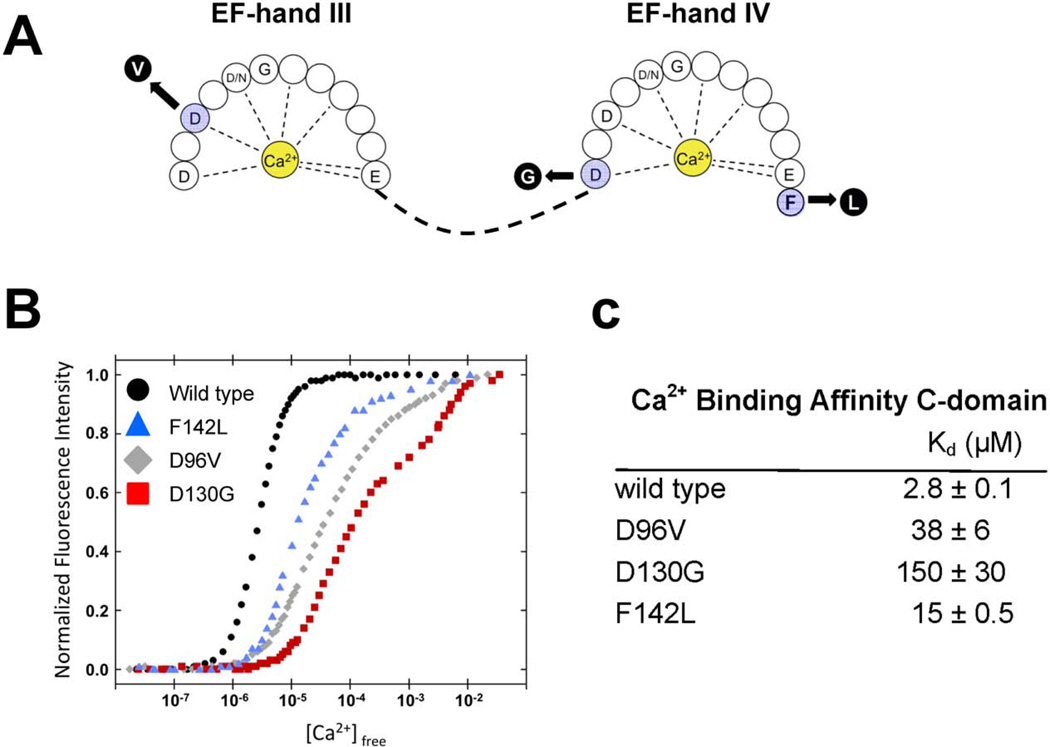
Methods and results: We ascertained 2 unrelated infants (probands) with recurrent cardiac arrest and dramatically prolonged QTc interval who were both born to healthy parents. The 2 parent-child trios were investigated with the use of exome sequencing to search for de novo genetic variants. We then performed follow-up candidate gene screening on an independent cohort of 82 subjects with congenital long-QT syndrome without an identified genetic cause. Biochemical studies were performed to determine the functional consequences of mutations discovered in 2 genes encoding calmodulin. We discovered 3 heterozygous de novo mutations in either CALM1 or CALM2, 2 of the 3 human genes encoding calmodulin, in the 2 probands and in 2 additional subjects with recurrent cardiac arrest. All mutation carriers were infants who exhibited life-threatening ventricular arrhythmias combined variably with epilepsy and delayed neurodevelopment. Mutations altered residues in or adjacent to critical calcium binding loops in the calmodulin carboxyl-terminal domain. Recombinant mutant calmodulins exhibited several-fold reductions in calcium binding affinity.
Conclusions: Human calmodulin mutations disrupt calcium ion binding to the protein and are associated with a life-threatening condition in early infancy. Defects in calmodulin function will disrupt important calcium signaling events in heart, affecting membrane ion channels, a plausible molecular mechanism for potentially deadly disturbances in heart rhythm during infancy.
Conflict of interest statement
Figures
Comment in
-
Using whole exome sequencing to walk from clinical practice to research and back again.Circulation. 2013 Mar 5;127(9):968-70. doi: 10.1161/CIRCULATIONAHA.113.001284. Epub 2013 Feb 6. Circulation. 2013. PMID: 23388216 No abstract available.
References
-
- Miller TE, Estrella E, Myerburg RJ, Garcia d V, Moreno N, Rusconi P, Ahearn ME, Baumbach L, Kurlansky P, Wolff G, Bishopric NH. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation. 2004;109:3029–3034. - PubMed
-
- Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, Richard TA, Berti MR, Bloise R. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343:262–267. - PubMed
-
- Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, Towbin JA. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–2269. - PubMed
-
- Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Rognum T, Schwartz PJ, George AL., Jr Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–376. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GGP09247/TI_/Telethon/Italy
- T32-NS007941/NS/NINDS NIH HHS/United States
- T32 NS007491/NS/NINDS NIH HHS/United States
- S10 RR028106/RR/NCRR NIH HHS/United States
- U01 HL065962/HL/NHLBI NIH HHS/United States
- R01 NS007941/NS/NINDS NIH HHS/United States
- K24 HL069712/HL/NHLBI NIH HHS/United States
- RR024975/RR/NCRR NIH HHS/United States
- RR028106/RR/NCRR NIH HHS/United States
- UL1 RR024975/RR/NCRR NIH HHS/United States
- HL083374/HL/NHLBI NIH HHS/United States
- R01 HL083374/HL/NHLBI NIH HHS/United States
- HL069712/HL/NHLBI NIH HHS/United States
- U19 HL065962/HL/NHLBI NIH HHS/United States
- HL065962/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
