

Branching process deconvolution algorithm reveals a detailed cell-cycle transcription program
- PMID: 23388635
- PMCID: PMC3593847
- DOI: 10.1073/pnas.1120991110
Branching process deconvolution algorithm reveals a detailed cell-cycle transcription program
Abstract
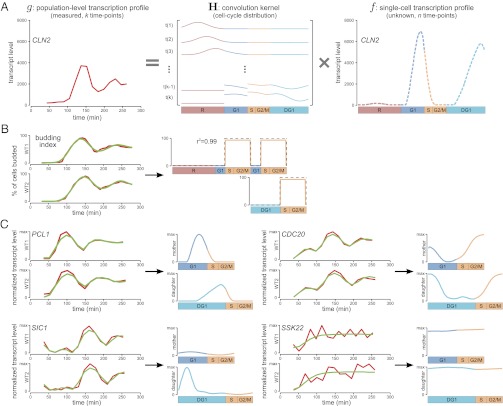
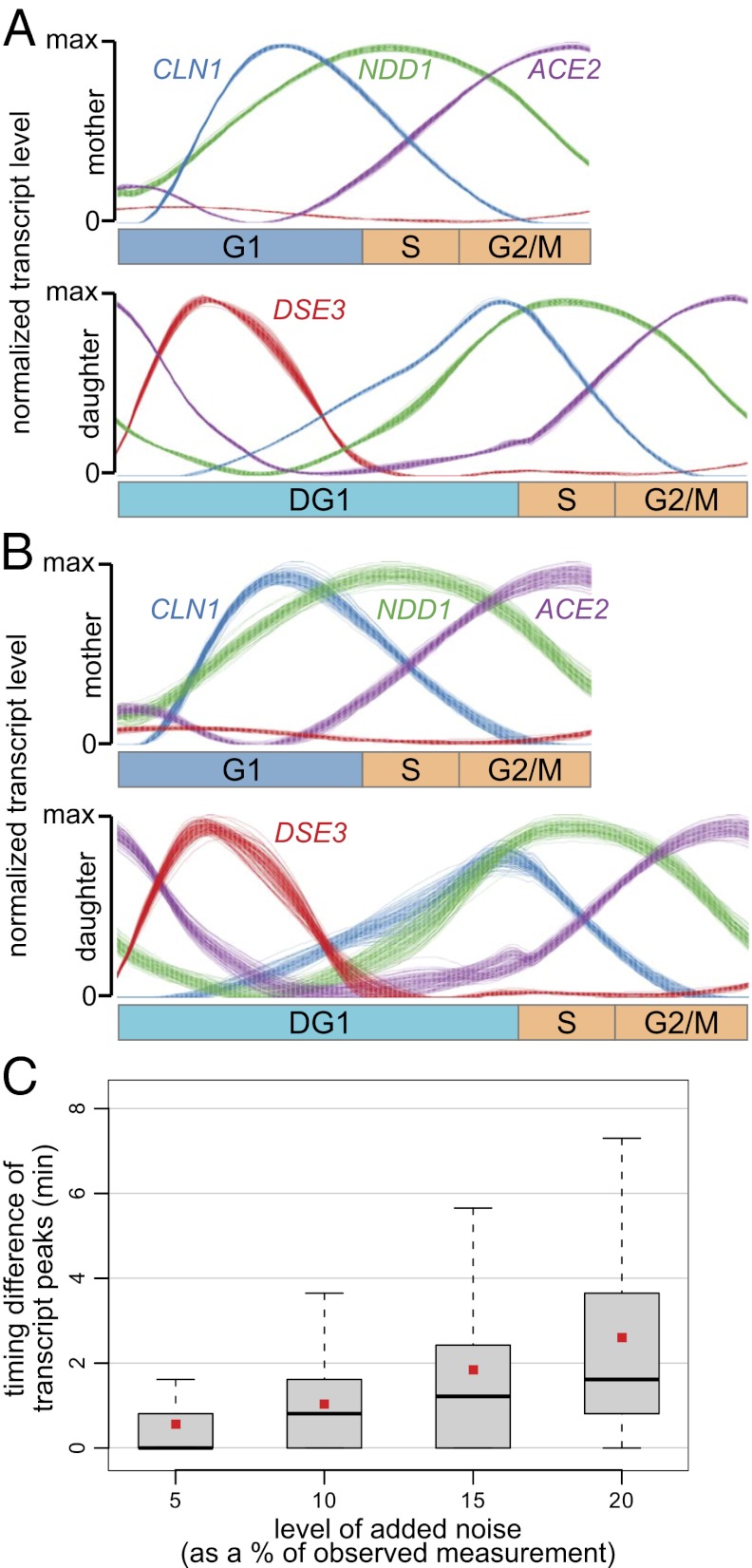
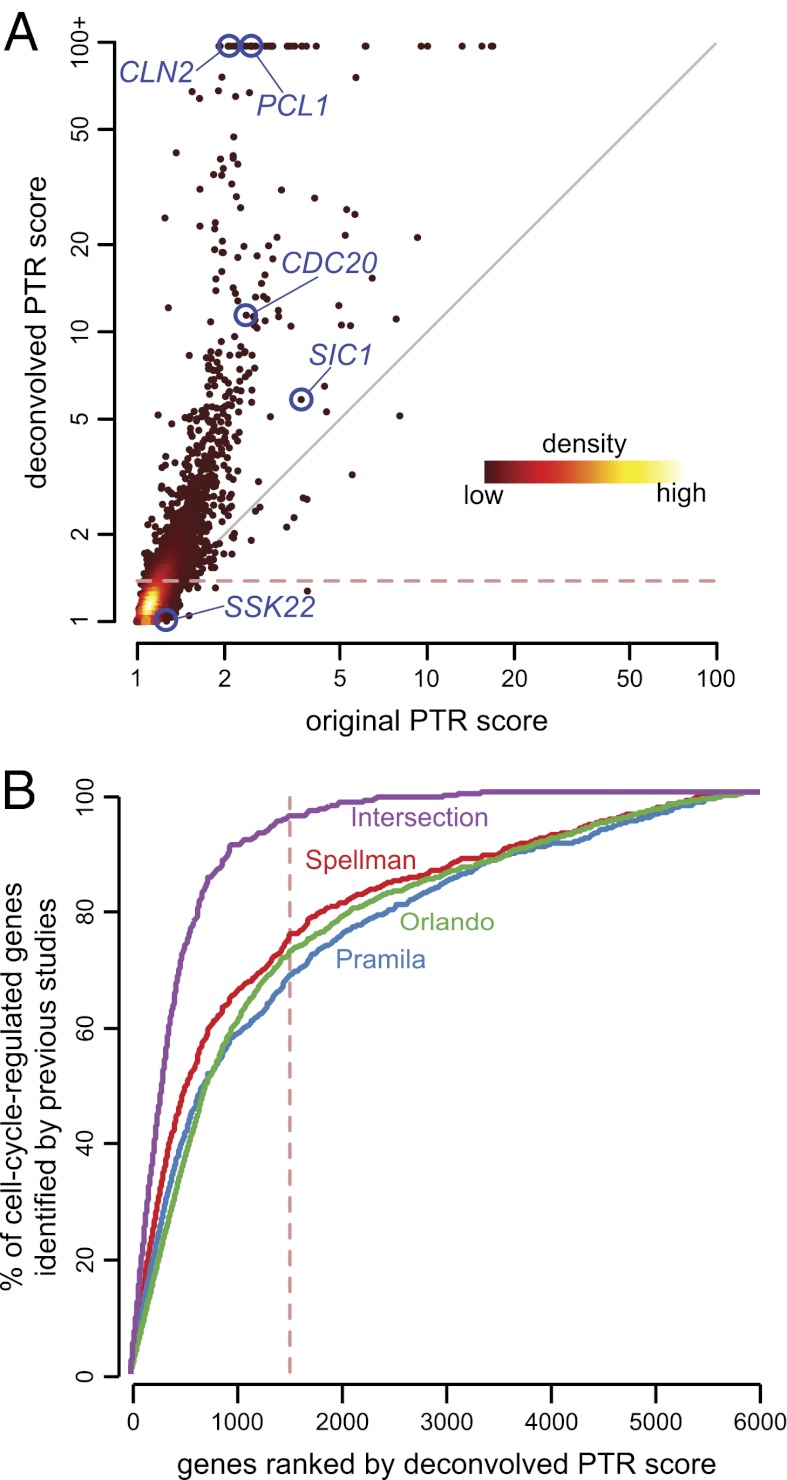
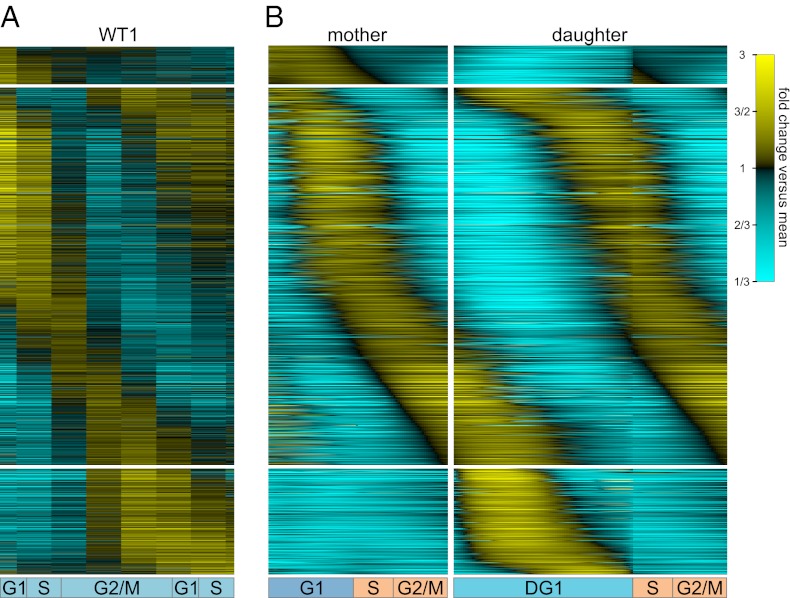
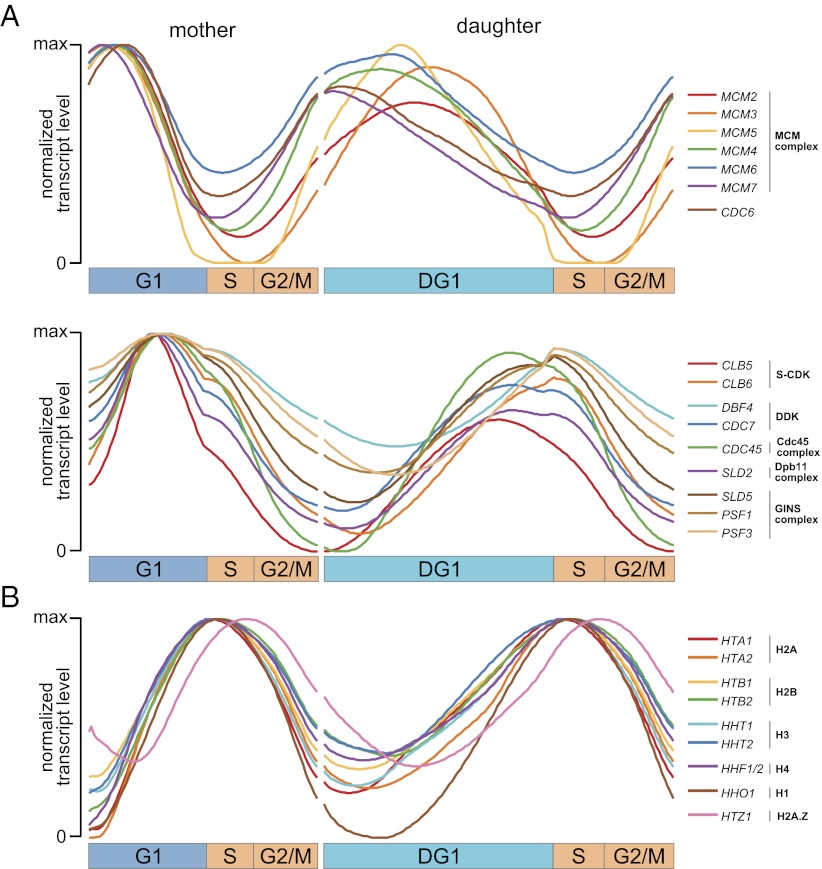
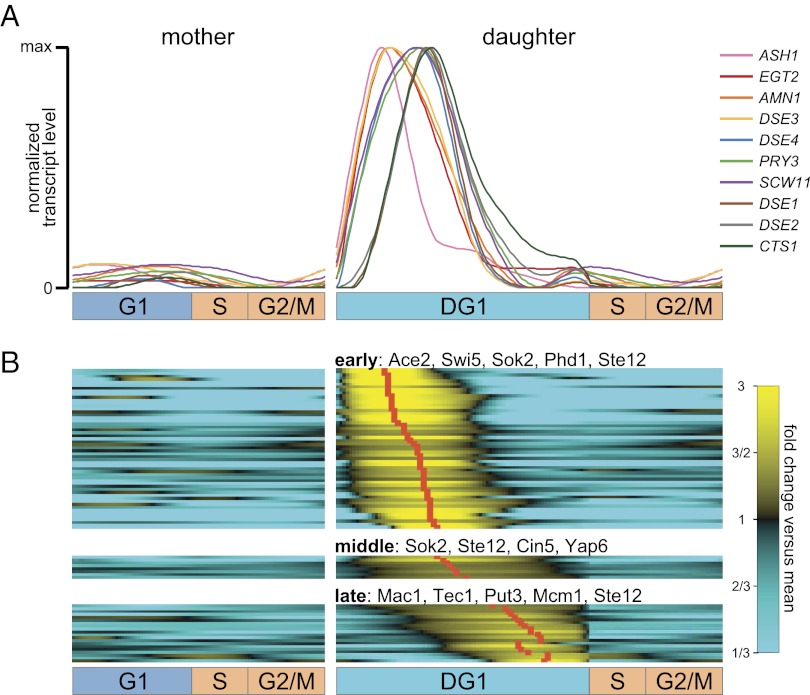
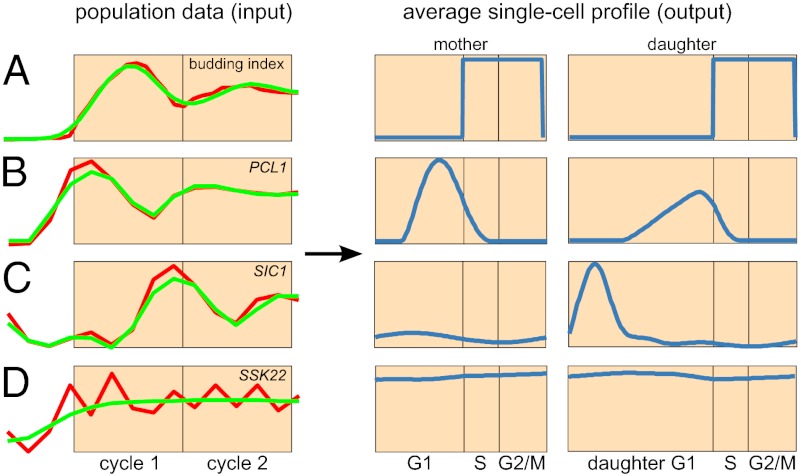
Due to cell-to-cell variability and asymmetric cell division, cells in a synchronized population lose synchrony over time. As a result, time-series measurements from synchronized cell populations do not reflect the underlying dynamics of cell-cycle processes. Here, we present a branching process deconvolution algorithm that learns a more accurate view of dynamic cell-cycle processes, free from the convolution effects associated with imperfect cell synchronization. Through wavelet-basis regularization, our method sharpens signal without sharpening noise and can remarkably increase both the dynamic range and the temporal resolution of time-series data. Although applicable to any such data, we demonstrate the utility of our method by applying it to a recent cell-cycle transcription time course in the eukaryote Saccharomyces cerevisiae. Our method more sensitively detects cell-cycle-regulated transcription and reveals subtle timing differences that are masked in the original population measurements. Our algorithm also explicitly learns distinct transcription programs for mother and daughter cells, enabling us to identify 82 genes transcribed almost entirely in early G1 in a daughter-specific manner.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
High-resolution timing of cell cycle-regulated gene expression.Proc Natl Acad Sci U S A. 2007 Oct 23;104(43):16892-7. doi: 10.1073/pnas.0706022104. Epub 2007 Sep 7. Proc Natl Acad Sci U S A. 2007. PMID: 17827275 Free PMC article.
-
Model-based deconvolution of cell cycle time-series data reveals gene expression details at high resolution.PLoS Comput Biol. 2009 Aug;5(8):e1000460. doi: 10.1371/journal.pcbi.1000460. Epub 2009 Aug 14. PLoS Comput Biol. 2009. PMID: 19680537 Free PMC article.
-
Different G1 cyclins control the timing of cell cycle commitment in mother and daughter cells of the budding yeast S. cerevisiae.Cell. 1992 Apr 17;69(2):317-27. doi: 10.1016/0092-8674(92)90412-6. Cell. 1992. PMID: 1533176
-
Cell cycle regulated transcription in yeast.Curr Opin Cell Biol. 1994 Jun;6(3):451-9. doi: 10.1016/0955-0674(94)90039-6. Curr Opin Cell Biol. 1994. PMID: 7917338 Review.
-
New cell cycle-regulated genes in the yeast Saccharomyces cerevisiae.Recent Results Cancer Res. 1997;143:251-61. doi: 10.1007/978-3-642-60393-8_18. Recent Results Cancer Res. 1997. PMID: 8912425 Review. No abstract available.
Cited by
-
Topology and control of the cell-cycle-regulated transcriptional circuitry.Genetics. 2014 Jan;196(1):65-90. doi: 10.1534/genetics.113.152595. Genetics. 2014. PMID: 24395825 Free PMC article. Review.
-
Connecting virulence pathways to cell-cycle progression in the fungal pathogen Cryptococcus neoformans.Curr Genet. 2017 Oct;63(5):803-811. doi: 10.1007/s00294-017-0688-5. Epub 2017 Mar 6. Curr Genet. 2017. PMID: 28265742 Free PMC article. Review.
-
Layers of regulation of cell-cycle gene expression in the budding yeast Saccharomyces cerevisiae.Mol Biol Cell. 2018 Nov 1;29(22):2644-2655. doi: 10.1091/mbc.E18-04-0255. Epub 2018 Sep 12. Mol Biol Cell. 2018. PMID: 30207828 Free PMC article.
-
A transcriptome-wide analysis deciphers distinct roles of G1 cyclins in temporal organization of the yeast cell cycle.Sci Rep. 2019 Mar 4;9(1):3343. doi: 10.1038/s41598-019-39850-7. Sci Rep. 2019. PMID: 30833602 Free PMC article.
-
Periodic mRNA synthesis and degradation co-operate during cell cycle gene expression.Mol Syst Biol. 2014 Jan 30;10(1):717. doi: 10.1002/msb.134886. Print 2014. Mol Syst Biol. 2014. PMID: 24489117 Free PMC article.
References
-
- Cho RJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2(1):65–73. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
