

Malaria biology and disease pathogenesis: insights for new treatments
- PMID: 23389616
- PMCID: PMC4783790
- DOI: 10.1038/nm.3073
Malaria biology and disease pathogenesis: insights for new treatments
Abstract
Plasmodium falciparum malaria, an infectious disease caused by a parasitic protozoan, claims the lives of nearly a million children each year in Africa alone and is a top public health concern. Evidence is accumulating that resistance to artemisinin derivatives, the frontline therapy for the asexual blood stage of the infection, is developing in southeast Asia. Renewed initiatives to eliminate malaria will benefit from an expanded repertoire of antimalarials, including new drugs that kill circulating P. falciparum gametocytes, thereby preventing transmission. Our current understanding of the biology of asexual blood-stage parasites and gametocytes and the ability to culture them in vitro lends optimism that high-throughput screenings of large chemical libraries will produce a new generation of antimalarial drugs. There is also a need for new therapies to reduce the high mortality of severe malaria. An understanding of the pathophysiology of severe disease may identify rational targets for drugs that improve survival.
Figures
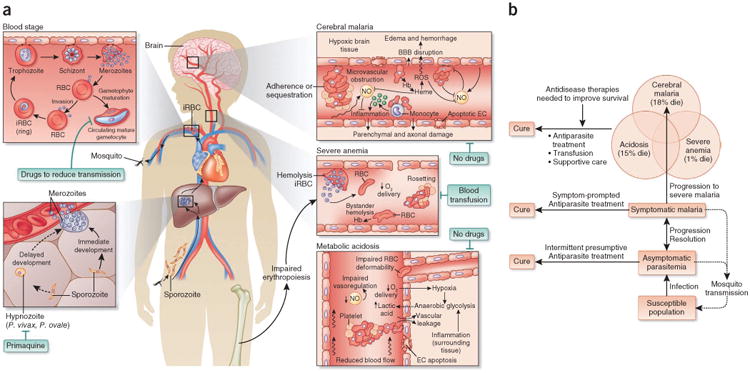
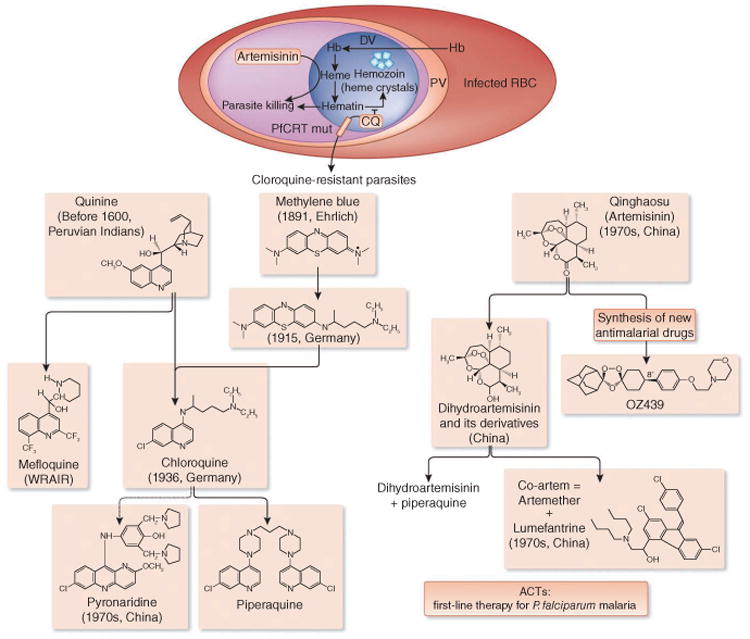
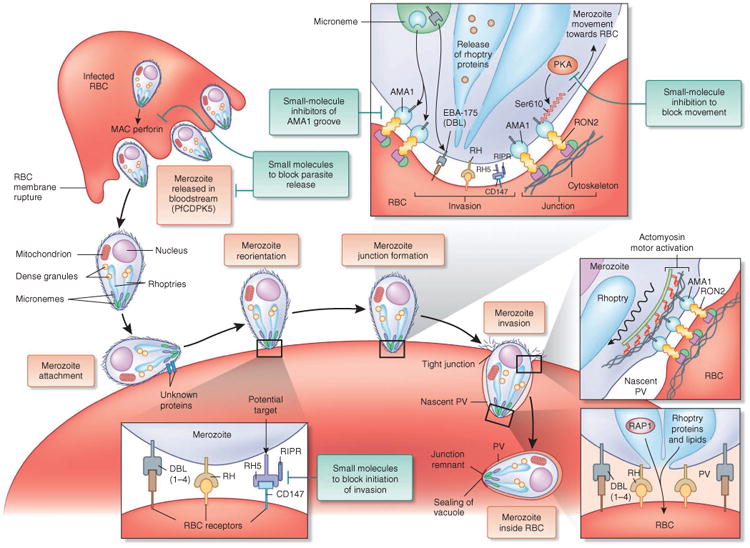
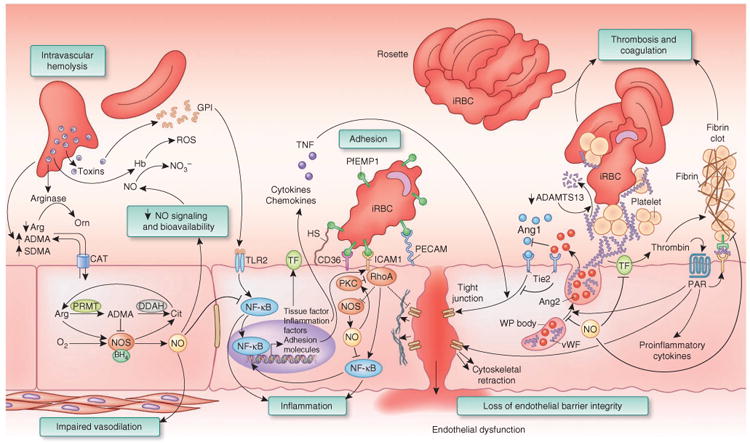
References
-
- Wells TN, Burrows JN, Baird JK. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 2010;26:145–151. - PubMed
-
- Burrows JN, Chibale K, Wells TN. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11:1226–1254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
