

Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity
- PMID: 23392653
- PMCID: PMC3683572
- DOI: 10.1161/CIRCGENETICS.112.964684
Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity
Abstract
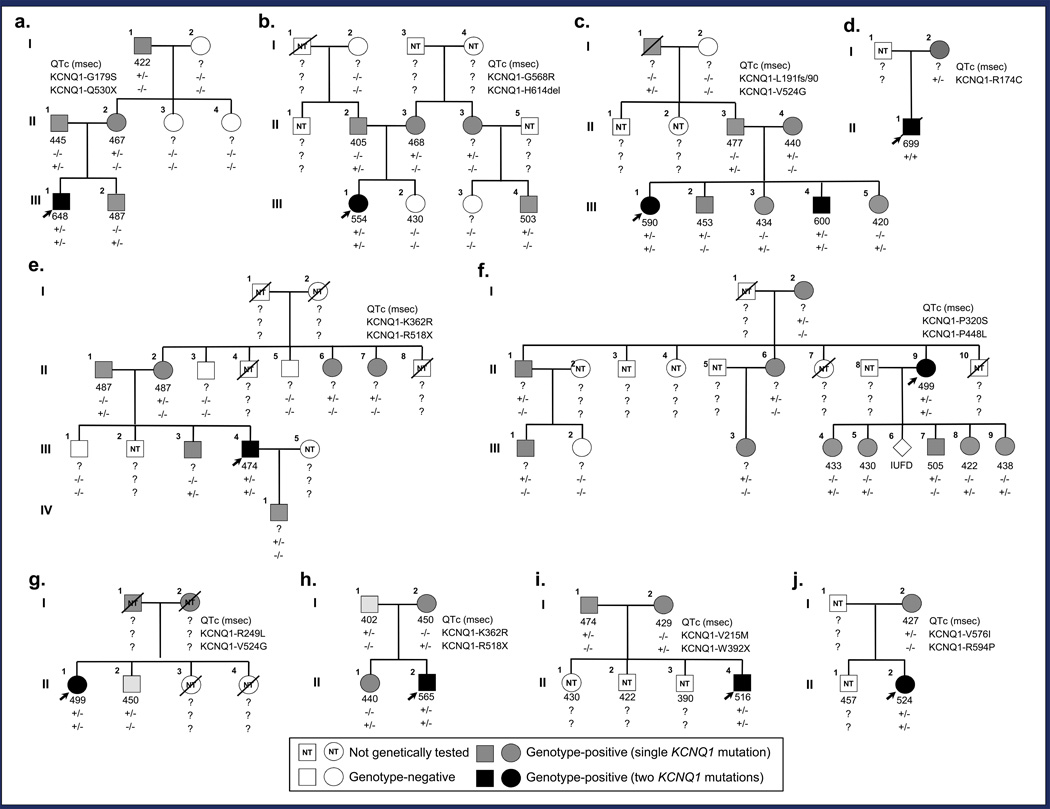
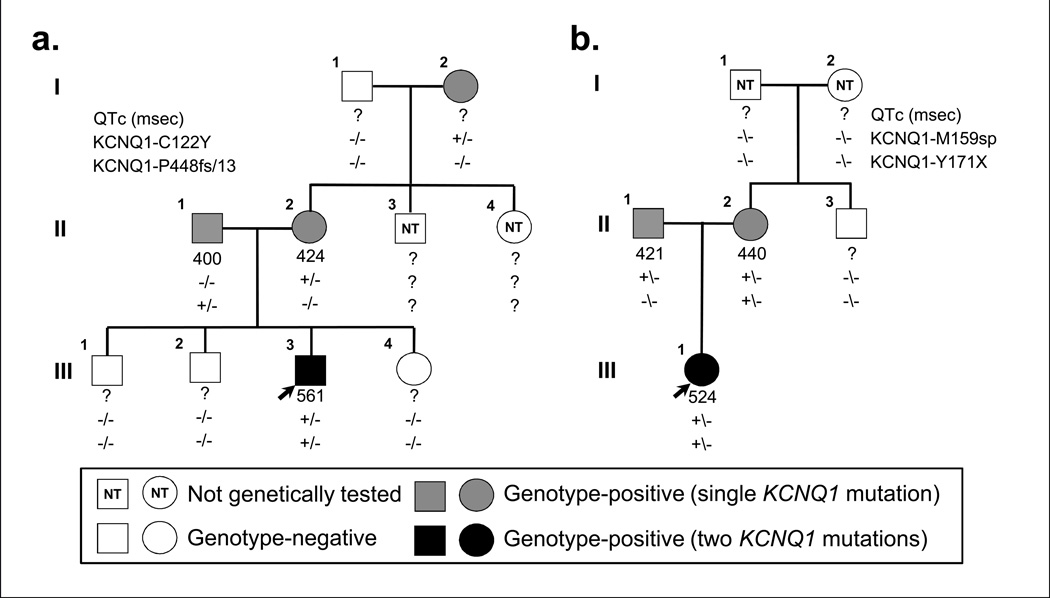
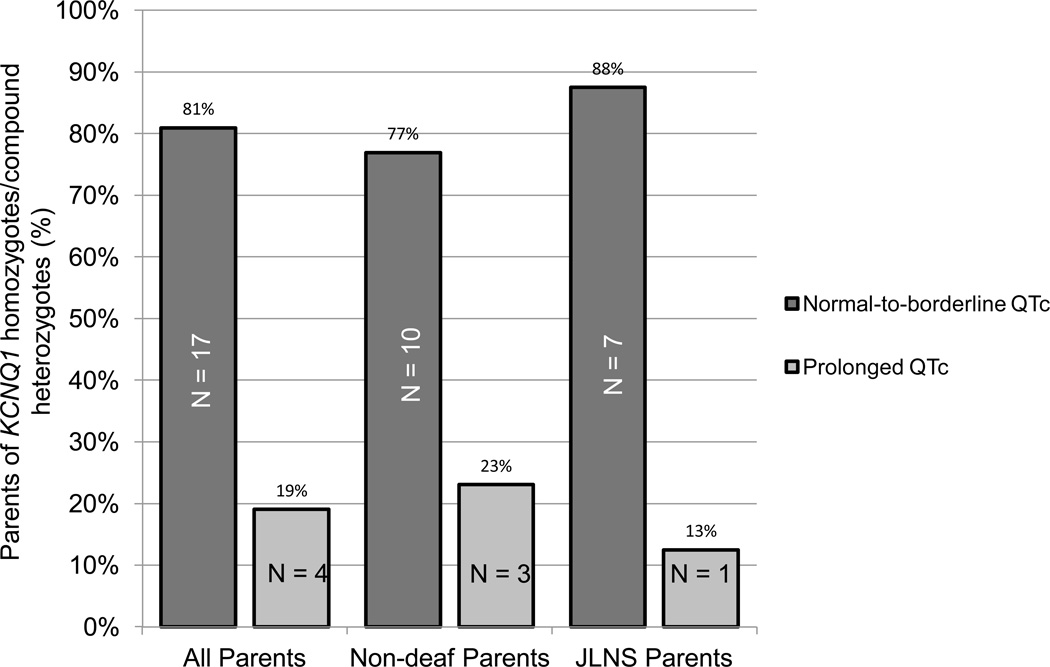
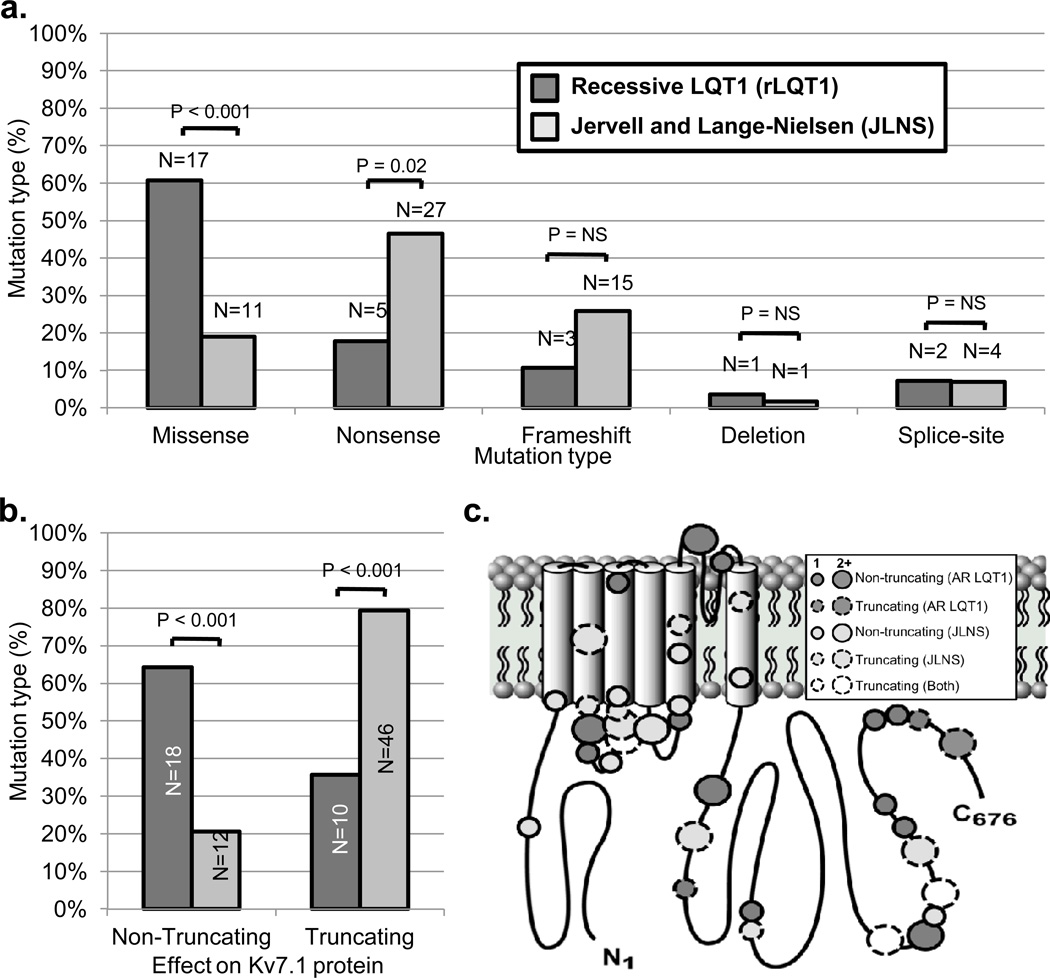
BACKGROUND- Homozygous or compound heterozygous mutations in KCNQ1 cause Jervell and Lange-Nielsen syndrome, a rare, autosomal-recessive form of long-QT syndrome characterized by deafness, marked QT prolongation, and a high risk of sudden death. However, it is not understood why some individuals with mutations on both KCNQ1 alleles present without deafness. In this study, we sought to determine the prevalence and genetic determinants of this phenomenon in a large referral population of patients with long-QT syndrome. METHODS AND RESULTS- A retrospective analysis of all patients with long-QT syndrome evaluated from July 1998 to April 2012 was used to identify those with ≥1 KCNQ1 mutation. Of the 249 KCNQ1-positive patients identified, 15 (6.0%) harbored a rare putative pathogenic mutation on both KCNQ1 alleles. Surprisingly, 11 of these patients (73%) presented without the sensorineural deafness associated with Jervell and Lange-Nielsen syndrome. The degree of QT-interval prolongation and the number of breakthrough cardiac events were similar between patients with and without deafness. Interestingly, truncating mutations were more prevalent in patients with Jervell and Lange-Nielsen syndrome (79%) than in nondeaf patients (36%; P<0.001) derived from this study and those in the literature. CONCLUSIONS- In this study, we provide evidence that the recessive inheritance of a severe long-QT syndrome type 1 phenotype in the absence of an auditory phenotype may represent a more common pattern of long-QT syndrome inheritance than previously anticipated and that these cases should be treated as a higher-risk long-QT syndrome subset similar to their Jervell and Lange-Nielsen syndrome counterparts. Furthermore, mutation type may serve as a genetic determinant of deafness, but not cardiac expressivity, in individuals harboring ≥1 KCNQ1 mutation on each allele.
Conflict of interest statement
Figures
Comment in
-
IKs in heart and hearing, the ear can do with less than the heart.Circ Cardiovasc Genet. 2013 Apr;6(2):141-3. doi: 10.1161/CIRCGENETICS.113.000143. Circ Cardiovasc Genet. 2013. PMID: 23591039 No abstract available.
References
-
- Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–2044. - PubMed
-
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. - PubMed
-
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. - PubMed
-
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
