

Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy
- PMID: 23393146
- PMCID: PMC3624668
- DOI: 10.1093/brain/aws343
Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy
Abstract
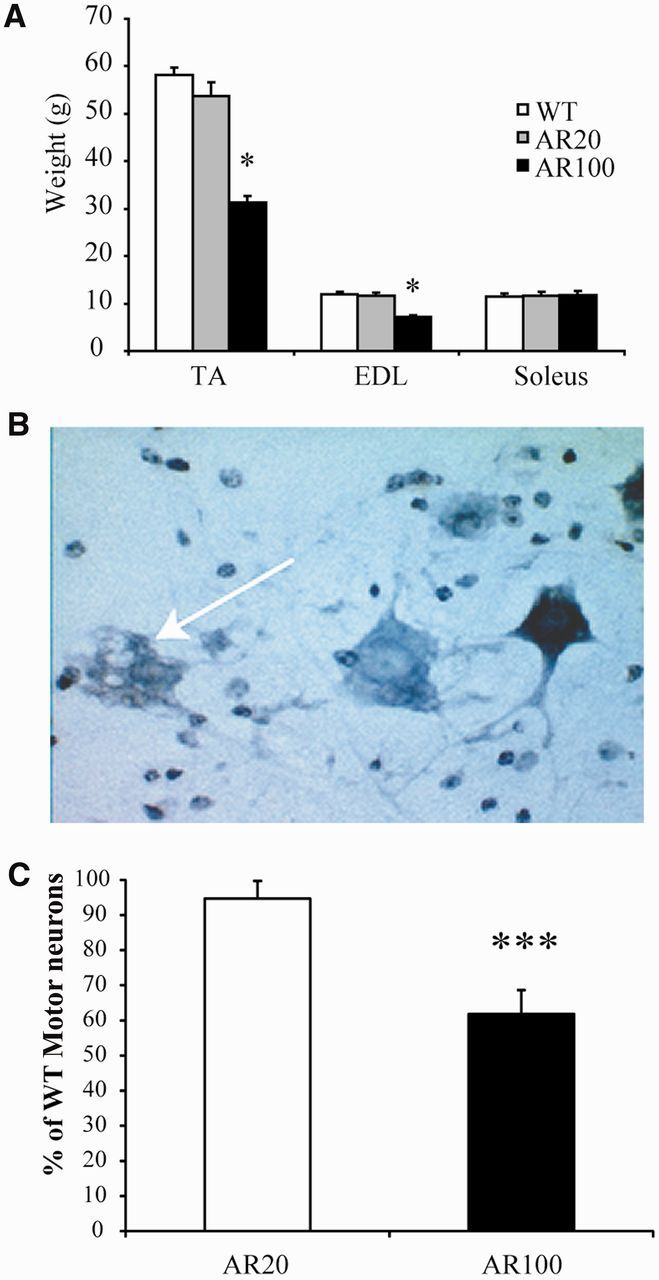
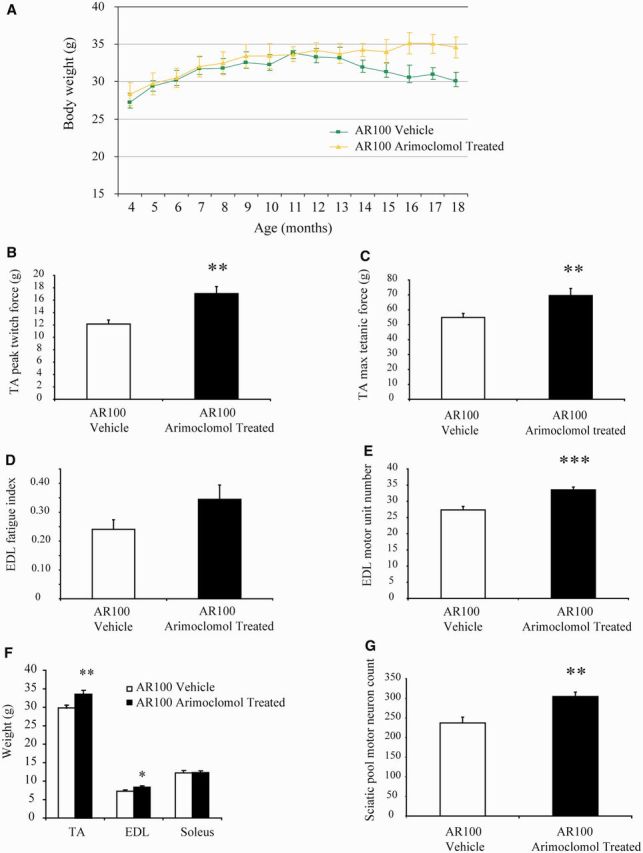
Spinal and bulbar muscular atrophy, also known as Kennedy's disease, is an adult-onset hereditary neurodegenerative disorder caused by an expansion of the polyglutamine repeat in the first exon in the androgen receptor gene. Pathologically, the disease is defined by selective loss of spinal and bulbar motor neurons causing bulbar, facial and limb weakness. Although the precise disease pathophysiology is largely unknown, it appears to be related to abnormal accumulation of the pathogenic androgen receptor protein within the nucleus, leading to disruption of cellular processes. Using a mouse model of spinal and bulbar muscular atrophy that exhibits many of the characteristic features of the human disease, in vivo physiological assessment of muscle function revealed that mice with the pathogenic expansion of the androgen receptor develop a motor deficit characterized by a reduction in muscle force, abnormal muscle contractile characteristics, loss of functional motor units and motor neuron degeneration. We have previously shown that treatment with arimoclomol, a co-inducer of the heat shock stress response, delays disease progression in the mutant superoxide dismutase 1 mouse model of amyotrophic lateral sclerosis, a fatal motor neuron disease. We therefore evaluated the therapeutic potential of arimoclomol in mice with spinal and bulbar muscular atrophy. Arimoclomol was administered orally, in drinking water, from symptom onset and the effects established at 18 months of age, a late stage of disease. Arimoclomol significantly improved hindlimb muscle force and contractile characteristics, rescued motor units and, importantly, improved motor neuron survival and upregulated the expression of the vascular endothelial growth factor which possess neurotrophic activity. These results provide evidence that upregulation of the heat shock response by treatment with arimoclomol may have therapeutic potential in the treatment of spinal and bulbar muscular atrophy and may also be a possible approach for the treatment of other neurodegenerative diseases.
Figures





Similar articles
-
The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol.Pharmacol Ther. 2014 Jan;141(1):40-54. doi: 10.1016/j.pharmthera.2013.08.003. Epub 2013 Aug 23. Pharmacol Ther. 2014. PMID: 23978556 Review.
-
Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models.Cell Stress Chaperones. 2020 Jan;25(1):173-191. doi: 10.1007/s12192-019-01064-1. Epub 2020 Jan 3. Cell Stress Chaperones. 2020. PMID: 31900865 Free PMC article.
-
Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy.Brain. 2014 Jul;137(Pt 7):1894-906. doi: 10.1093/brain/awu114. Epub 2014 Jun 4. Brain. 2014. PMID: 24898351 Free PMC article.
-
Arimoclomol, a coinducer of heat shock proteins for the potential treatment of amyotrophic lateral sclerosis.IDrugs. 2010 Jul;13(7):482-96. IDrugs. 2010. PMID: 20582873 Review.
-
Deterioration of muscle force and contractile characteristics are early pathological events in spinal and bulbar muscular atrophy mice.Dis Model Mech. 2020 May 26;13(5):dmm042424. doi: 10.1242/dmm.042424. Dis Model Mech. 2020. PMID: 32152060 Free PMC article.
Cited by
-
MiR-206 Attenuates Denervation-Induced Skeletal Muscle Atrophy in Rats Through Regulation of Satellite Cell Differentiation via TGF-β1, Smad3, and HDAC4 Signaling.Med Sci Monit. 2016 Apr 7;22:1161-70. doi: 10.12659/msm.897909. Med Sci Monit. 2016. PMID: 27054781 Free PMC article.
-
Molecular chaperones and protein folding as therapeutic targets in Parkinson's disease and other synucleinopathies.Acta Neuropathol Commun. 2013 Dec 5;1(1):79. doi: 10.1186/2051-5960-1-79. Acta Neuropathol Commun. 2013. PMID: 24314025 Free PMC article. Review.
-
HSF1 and Its Role in Huntington's Disease Pathology.Adv Exp Med Biol. 2023;1410:35-95. doi: 10.1007/5584_2022_742. Adv Exp Med Biol. 2023. PMID: 36396925 Free PMC article. Review.
-
Therapeutic targeting of the polyglutamine androgen receptor in Spinal and Bulbar Muscular Atrophy.Expert Opin Ther Targets. 2025 Jan-Feb;29(1-2):29-41. doi: 10.1080/14728222.2025.2464173. Epub 2025 Feb 10. Expert Opin Ther Targets. 2025. PMID: 39915972 Review.
-
Heat increases full-length SMN splicing: promise for splice-augmenting therapies for SMA.Hum Genet. 2022 Feb;141(2):239-256. doi: 10.1007/s00439-021-02408-7. Epub 2022 Jan 28. Hum Genet. 2022. PMID: 35088120 Free PMC article.
References
-
- Abel A, Walcott J, Woods J, Duda J, Merry DE. Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum Mol Genet. 2001;10:107–16. - PubMed
-
- Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Do J, et al. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10:1039–48. - PubMed
-
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, et al. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–11. - PMC - PubMed
-
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
