

Mutations in MED12 cause X-linked Ohdo syndrome
- PMID: 23395478
- PMCID: PMC3591845
- DOI: 10.1016/j.ajhg.2013.01.007
Mutations in MED12 cause X-linked Ohdo syndrome
Abstract
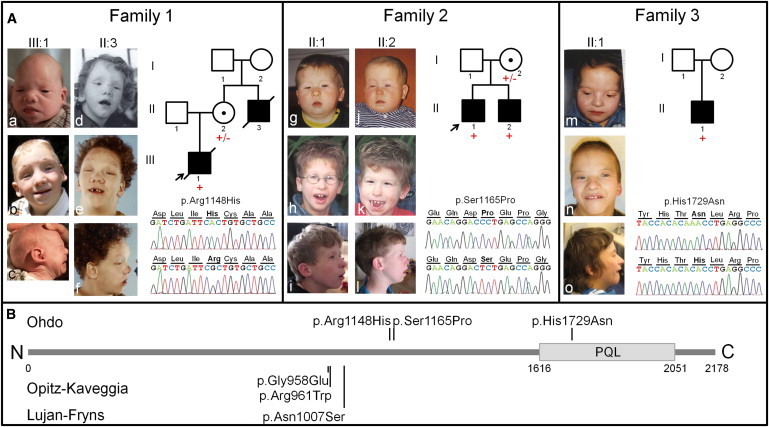
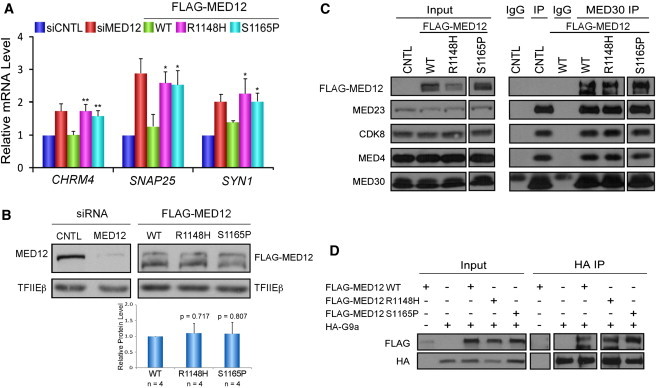
Ohdo syndrome comprises a heterogeneous group of disorders characterized by intellectual disability (ID) and typical facial features, including blepharophimosis. Clinically, these blepharophimosis-ID syndromes have been classified in five distinct subgroups, including the Maat-Kievit-Brunner (MKB) type, which, in contrast to the others, is characterized by X-linked inheritance and facial coarsening at older age. We performed exome sequencing in two families, each with two affected males with Ohdo syndrome MKB type. In the two families, MED12 missense mutations (c.3443G>A [p.Arg1148His] or c.3493T>C [p.Ser1165Pro]) segregating with the phenotype were identified. Upon subsequent analysis of an additional cohort of nine simplex male individuals with Ohdo syndrome, one additional de novo missense change (c.5185C>A [p.His1729Asn]) in MED12 was detected. The occurrence of three different hemizygous missense mutations in three unrelated families affected by Ohdo syndrome MKB type shows that mutations in MED12 are the underlying cause of this X-linked form of Ohdo syndrome. Together with the recently described KAT6B mutations resulting in Ohdo syndrome Say/Barber/Biesecker/Young/Simpson type, our findings point to aberrant chromatin modification as being central to the pathogenesis of Ohdo syndrome.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Verloes A., Bremond-Gignac D., Isidor B., David A., Baumann C., Leroy M.A., Stevens R., Gillerot Y., Héron D., Héron B. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A. 2006;140:1285–1296. - PubMed
-
- Day R., Beckett B., Donnai D., Fryer A., Heidenblad M., Howard P., Kerr B., Mansour S., Maye U., McKee S. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008;74:434–444. - PubMed
-
- Clayton-Smith J., O’Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A.K., Thomas T., Biesecker L.G., Smith P. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011;89:675–681. - PMC - PubMed
-
- Kim S., Xu X., Hecht A., Boyer T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006;281:14066–14075. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
