

Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition
- PMID: 23396353
- PMCID: PMC3897332
- DOI: 10.1038/nsmb.2501
Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition
Abstract
Topoisomerase I (TOP1) inhibitors are an important class of anticancer drugs. The cytotoxicity of TOP1 inhibitors can be modulated by replication fork reversal through a process that requires poly(ADP-ribose) polymerase (PARP) activity. Whether regressed forks can efficiently restart and what factors are required to restart fork progression after fork reversal are still unknown. We have combined biochemical and EM approaches with single-molecule DNA fiber analysis to identify a key role for human RECQ1 helicase in replication fork restart after TOP1 inhibition that is not shared by other human RecQ proteins. We show that the poly(ADP-ribosyl)ation activity of PARP1 stabilizes forks in the regressed state by limiting their restart by RECQ1. These studies provide new mechanistic insights into the roles of RECQ1 and PARP in DNA replication and offer molecular perspectives to potentiate chemotherapeutic regimens based on TOP1 inhibition.
Figures
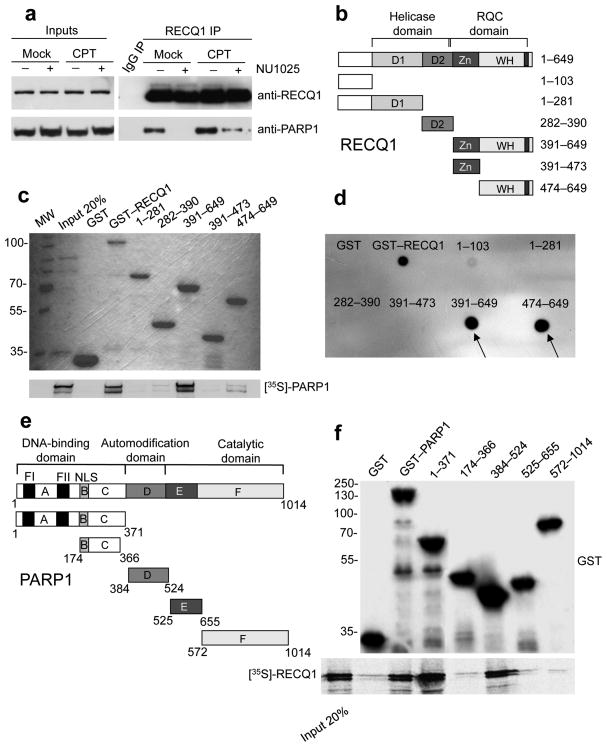
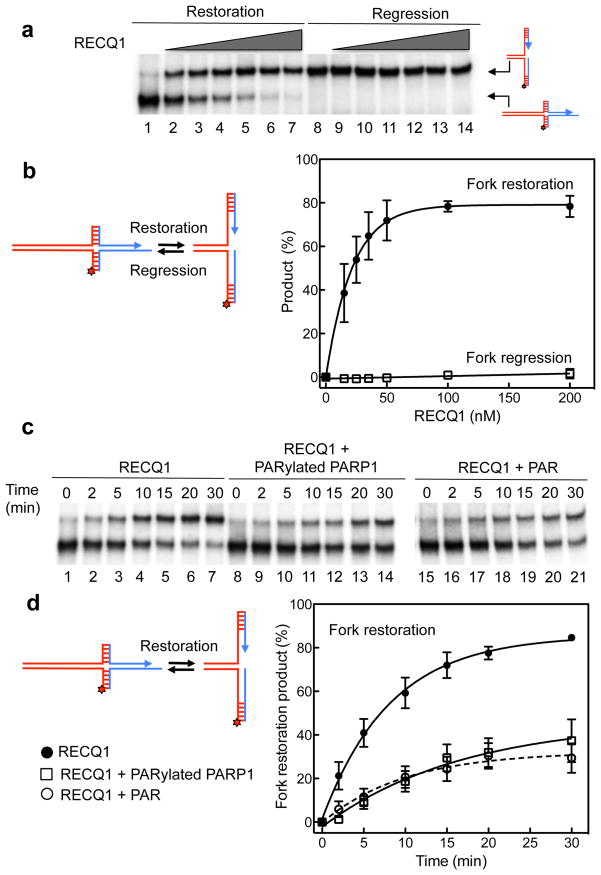
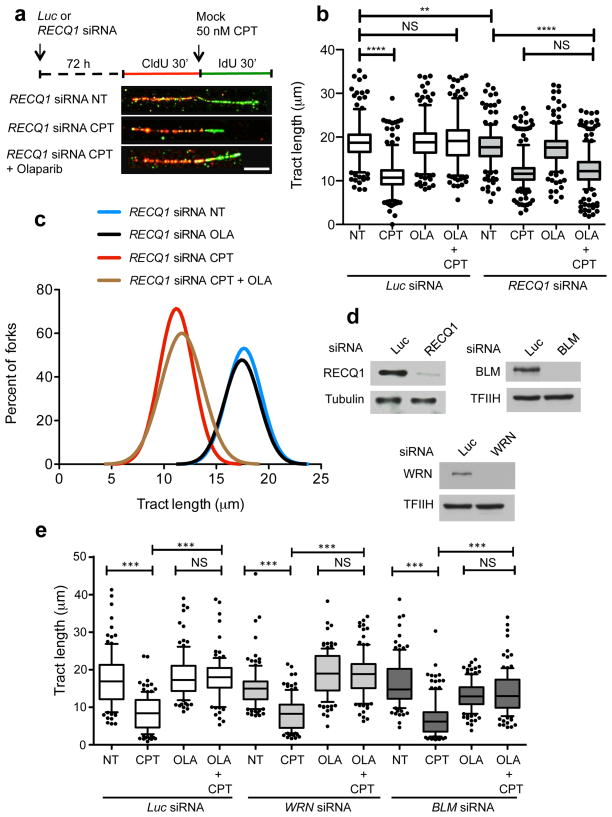
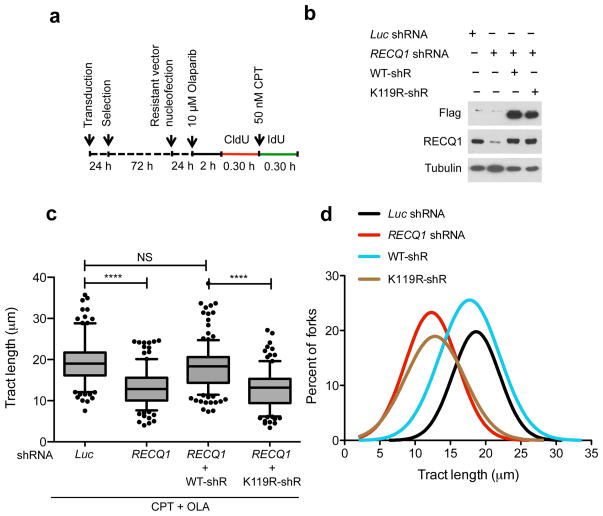
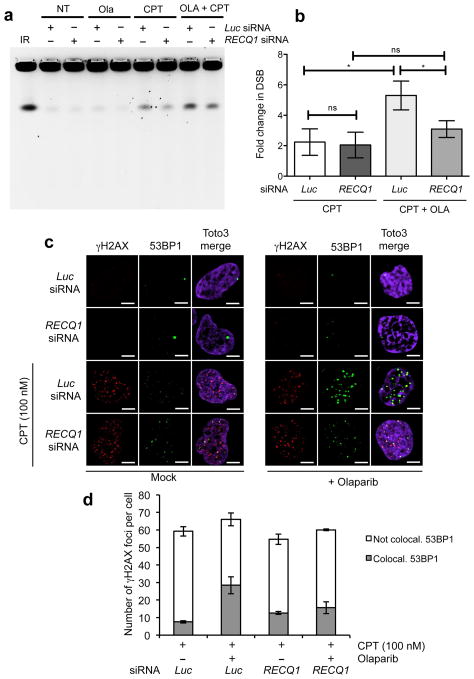
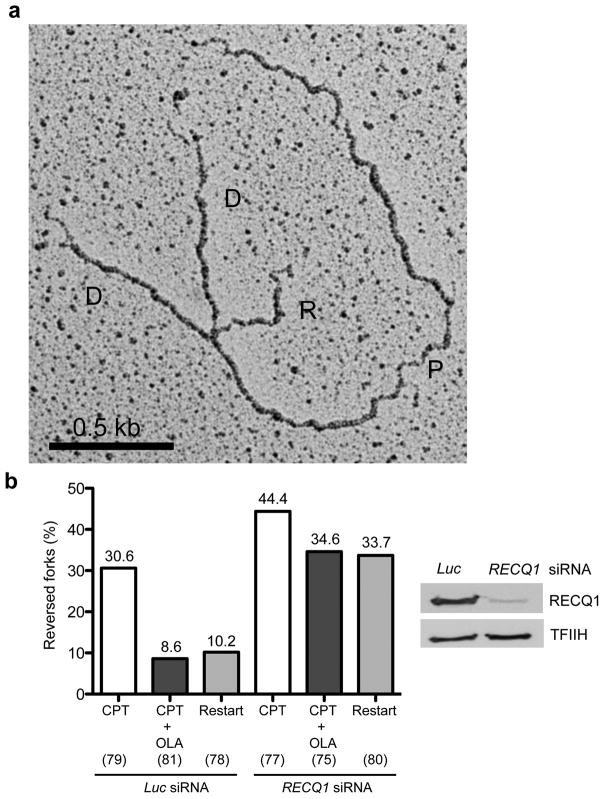
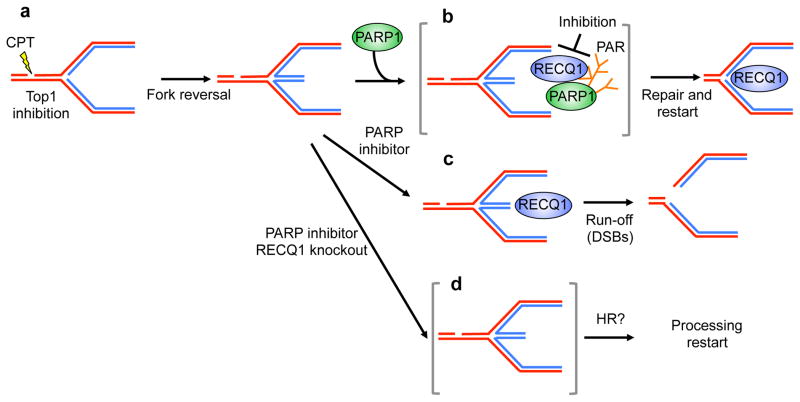
References
-
- Koster DA, Palle K, Bot ES, Bjornsti MA, Dekker NH. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature. 2007;448:213–7. - PubMed
-
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature reviews Cancer. 2006;6:789–802. - PubMed
-
- Rodriguez-Galindo C, et al. Clinical use of topoisomerase I inhibitors in anticancer treatment. Medical and pediatric oncology. 2000;35:385–402. - PubMed
-
- Pommier Y, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutation research. 2003;532:173–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
