

The multifaceted role of mTORC1 in the control of lipid metabolism
- PMID: 23399656
- PMCID: PMC3589096
- DOI: 10.1038/embor.2013.5
The multifaceted role of mTORC1 in the control of lipid metabolism
Abstract
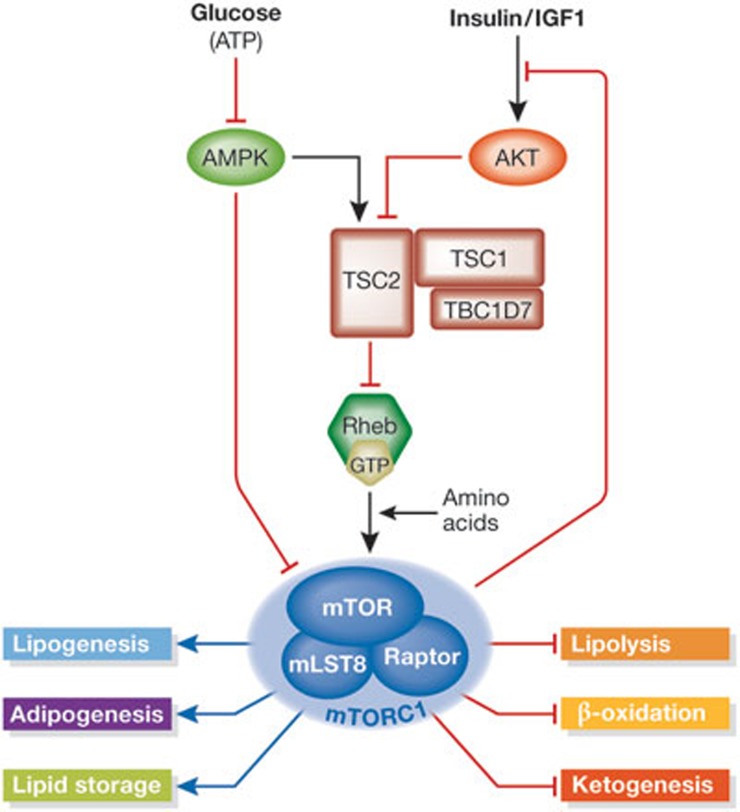
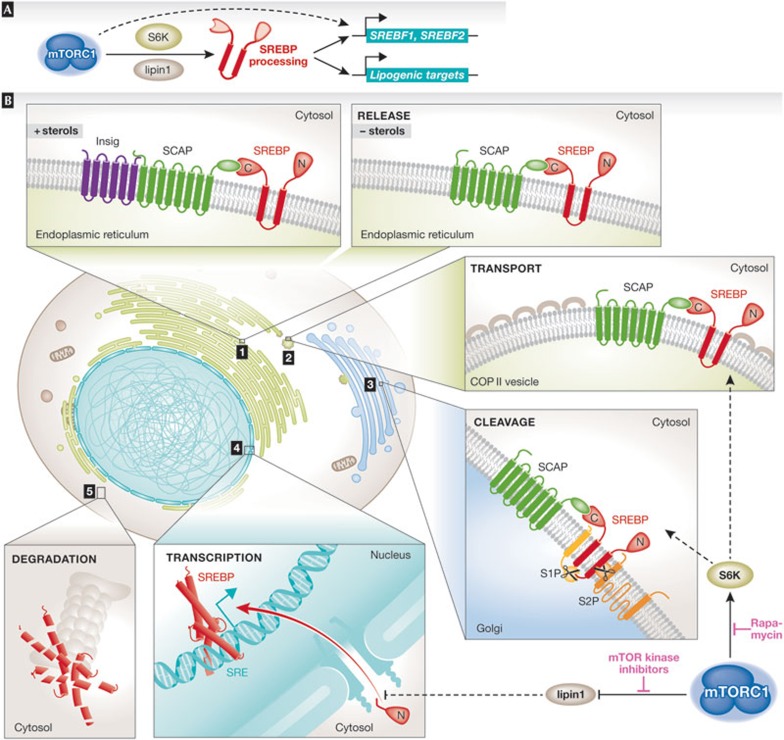
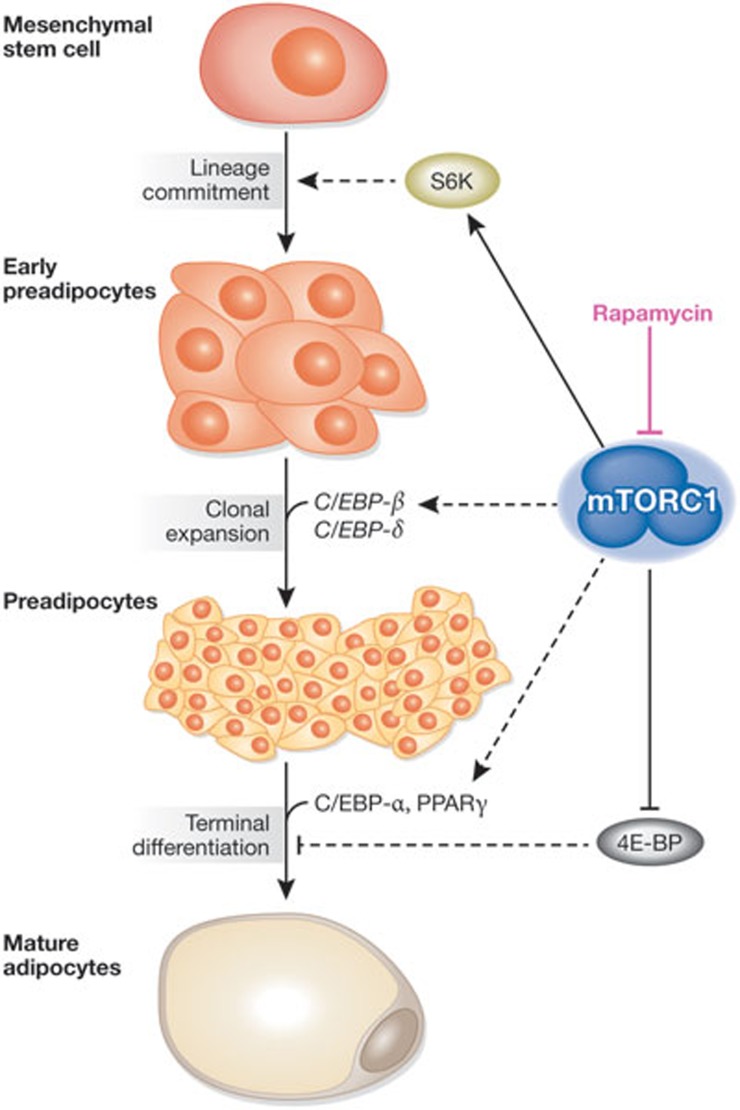
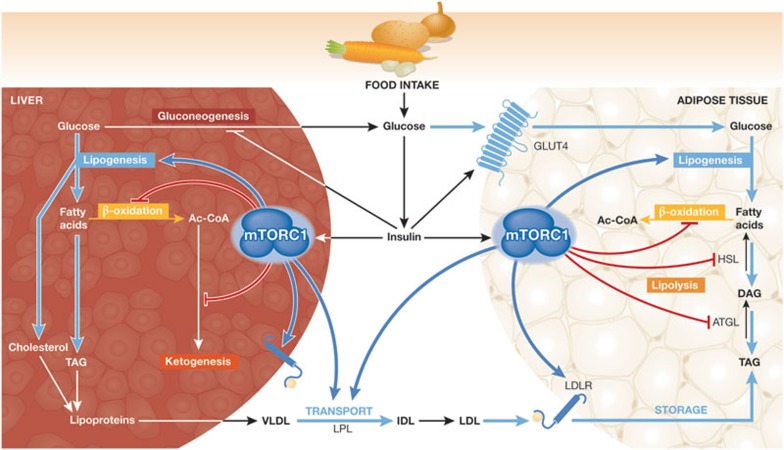
The mechanistic target of rapamycin is a protein kinase that, as part of the mechanistic target of rapamycin complex 1 (mTORC1), senses both local nutrients and, through insulin signalling, systemic nutrients to control a myriad of cellular processes. Although roles for mTORC1 in promoting protein synthesis and inhibiting autophagy in response to nutrients have been well established, it is emerging as a central regulator of lipid homeostasis. Here, we discuss the growing genetic and pharmacological evidence demonstrating the functional importance of its signalling in controlling mammalian lipid metabolism, including lipid synthesis, oxidation, transport, storage and lipolysis, as well as adipocyte differentiation and function. Defining the role of mTORC1 signalling in these metabolic processes is crucial to understanding the pathophysiology of obesity and its relationship to complex diseases, including diabetes and cancer.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 - PubMed
-
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162 - PubMed
-
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
