

Comment
doi: 10.1158/1078-0432.CCR-13-0051.
Epub 2013 Feb 12.
Glioblastoma resistance to anti-VEGF therapy: has the challenge been MET?
Affiliations
- PMID: 23403631
- PMCID: PMC3618531
- DOI: 10.1158/1078-0432.CCR-13-0051
Item in Clipboard
Comment
Glioblastoma resistance to anti-VEGF therapy: has the challenge been MET?
Clin Cancer Res.
.
Abstract
In glioblastoma cells the receptor tyrosine kinase c-Met is upregulated in response to bevacizumab and plays an important role in promoting invasion and tumor recurrence. These data support novel links between VEGF-A and hepatocyte growth factor and suggest that c-Met and its signaling effectors may be effective targets for anti-invasive therapies.
©2013 AACR.
Conflict of interest statement
The author declares no conflicts of interest.
Figures
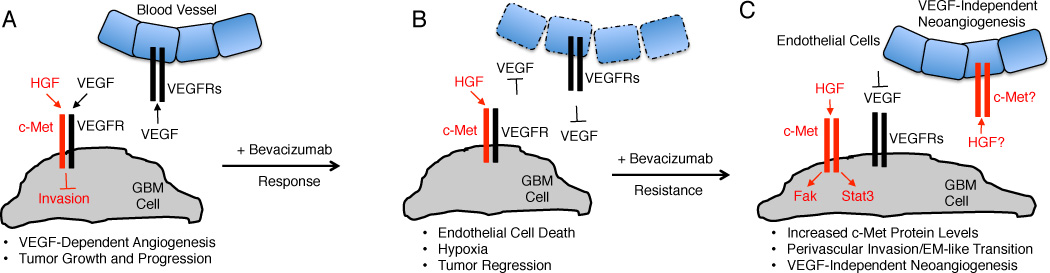
(A); Tumor cell proliferation and invasion are regulated via cross talk between the VEGF-A and HGF signaling pathways. During GB growth and progression c-Met and VEGFR-2 form heterodimeric complexes that suppress cell invasion via PTP1B-mediated Met tyrosine dephosphorylation. Meanwhile, VEGF-A produced by GB cells interacts with its receptors in endothelial cells and promotes angiogenesis and blood vessel permeability in the vascular niche. (B); Bevacizumab neutralizes VEGF-A signaling, leading to endothelial cell death, tumor regression, and transient improvements in overall and progression-free survival. (C); Responsiveness to Bevacizumab is followed by resistance, due to upregulation of c-Met gene expression in GB cells (acquired resistance) or selective survival of sub-populations of tumor cells that overexpress c-Met (intrinsic resistance). Met forms homodimers that bind HGF/SF and via an autocrine signaling loop promote Stat3 and Fak phosphorylation, promoting a mesenchymal-like phenotypes and perivascular tumor cell invasion. In Bevacizumab resistant tumors, paracrine HGF/SF signaling via Met in endothelial cells may also promote VEGF-independent neoangiogenesis.
Comment on
-
Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance.Clin Cancer Res. 2013 Apr 1;19(7):1773-83. doi: 10.1158/1078-0432.CCR-12-1281. Epub 2013 Jan 10. Clin Cancer Res. 2013. PMID: 23307858 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
