

Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability
- PMID: 23403903
- PMCID: PMC3778355
- DOI: 10.1038/ejhg.2013.17
Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability
Abstract
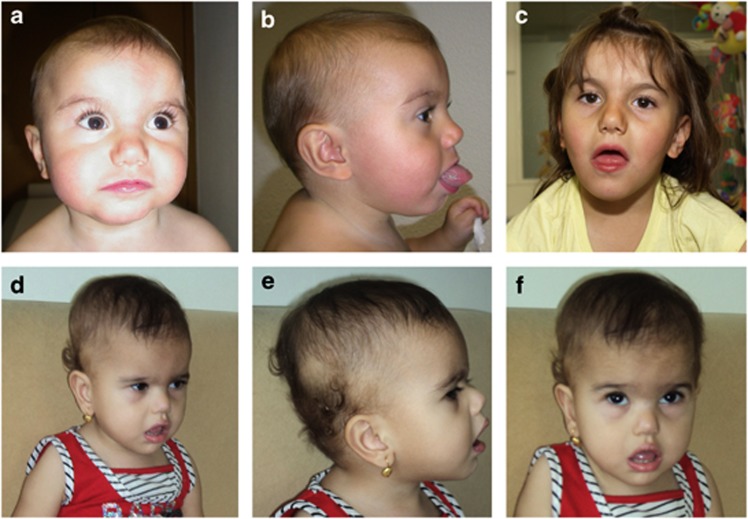
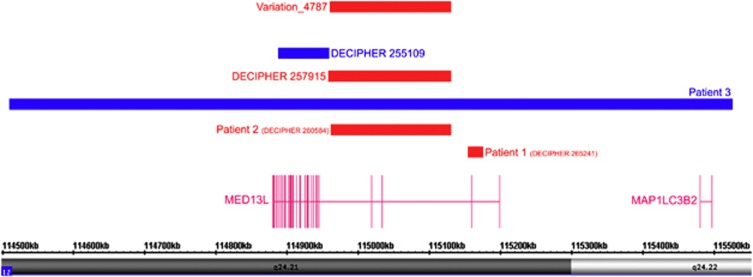
A chromosomal balanced translocation disrupting the MED13L (Mediator complex subunit13-like) gene, encoding a subunit of the Mediator complex, was previously associated with transposition of the great arteries (TGA) and intellectual disability (ID), and led to the identification of missense mutations in three patients with isolated TGA. Recently, a homozygous missense mutation in MED13L was found in two siblings with non-syndromic ID from a consanguineous family. Here, we describe for the first time, three patients with copy number changes affecting MED13L and delineate a recognizable MED13L haploinsufficiency syndrome. Using high resolution molecular karyotyping, we identified two intragenic de novo frameshift deletions, likely resulting in haploinsufficiency, in two patients with a similar phenotype of hypotonia, moderate ID, conotruncal heart defect and facial anomalies. In both, Sanger sequencing of MED13L did not reveal any pathogenic mutation and exome sequencing in one patient showed no evidence for a non-allelic second hit. A further patient with hypotonia, learning difficulties and perimembranous VSD showed a 1 Mb de novo triplication in 12q24.2, including MED13L and MAP1LC3B2. Our findings show that MED13L haploinsufficiency in contrast to the previously observed missense mutations cause a distinct syndromic phenotype. Additionally, a MED13L copy number gain results in a milder phenotype. The clinical features suggesting a neurocristopathy may be explained by animal model studies indicating involvement of the Mediator complex subunit 13 in neural crest induction.
Figures
References
-
- Sato S, Tomomori-Sato C, Parmely TJ, et al. A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol Cell. 2004;14:685–691. - PubMed
-
- Muncke N, Jung C, Rudiger H, et al. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries) Circulation. 2003;108:2843–2850. - PubMed
-
- Musante L, Bartsch O, Ropers HH, Kalscheuer VM. cDNA cloning and characterization of the human THRAP2 gene which maps to chromosome 12q24, and its mouse ortholog Thrap2. Gene. 2004;332:119–127. - PubMed
-
- Risheg H, Graham JM, Clark RD, et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet. 2007;39:451–453. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
