

Congenital myasthenic syndromes due to mutations in ALG2 and ALG14
- PMID: 23404334
- PMCID: PMC3580273
- DOI: 10.1093/brain/awt010
Congenital myasthenic syndromes due to mutations in ALG2 and ALG14
Abstract
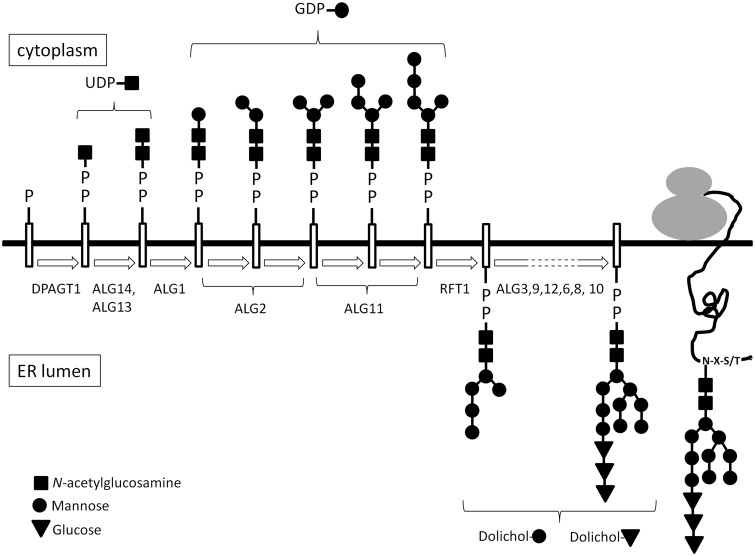
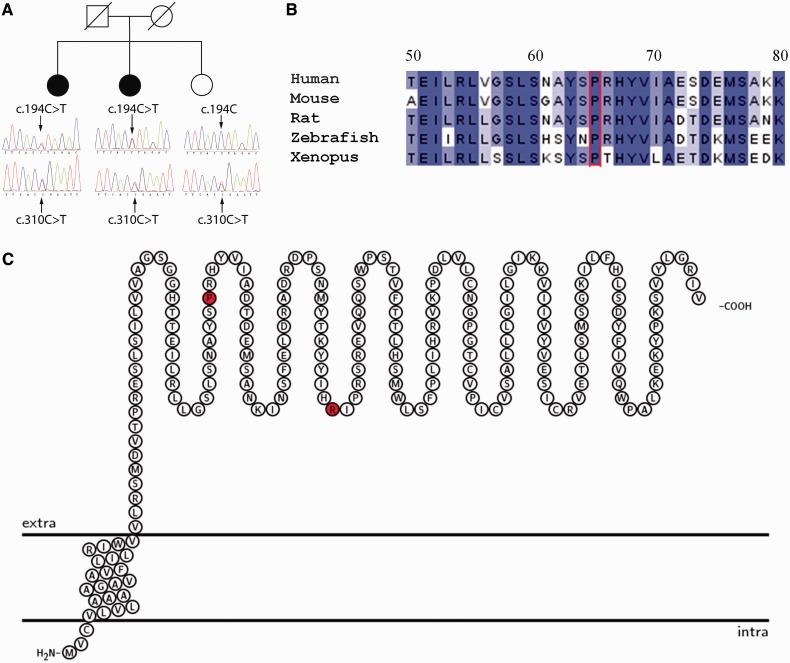
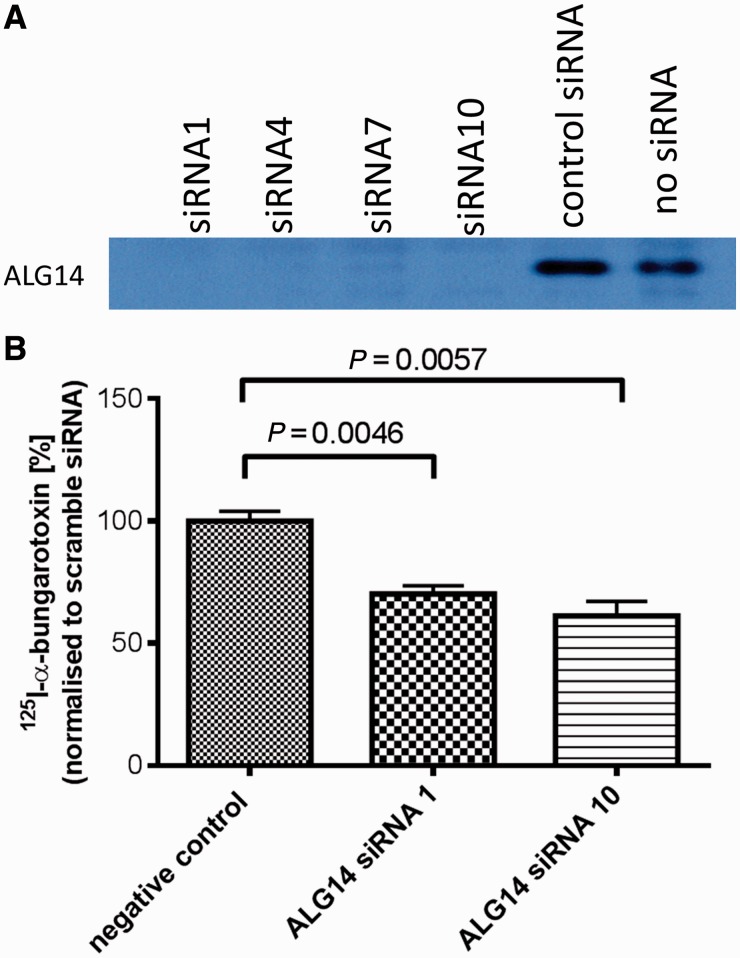
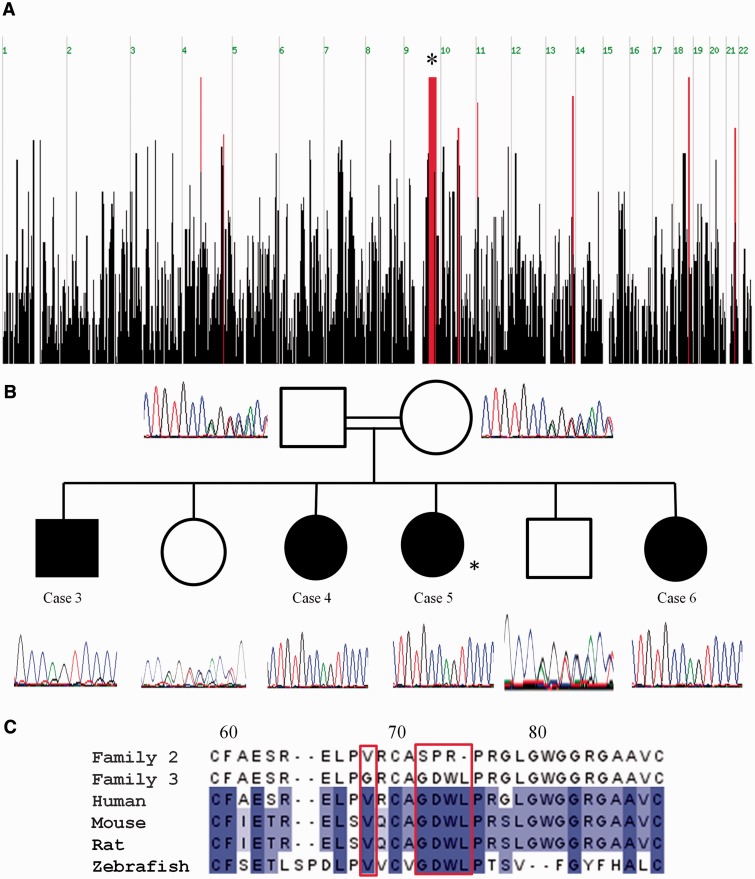
Congenital myasthenic syndromes are a heterogeneous group of inherited disorders that arise from impaired signal transmission at the neuromuscular synapse. They are characterized by fatigable muscle weakness. We performed linkage analysis, whole-exome and whole-genome sequencing to determine the underlying defect in patients with an inherited limb-girdle pattern of myasthenic weakness. We identify ALG14 and ALG2 as novel genes in which mutations cause a congenital myasthenic syndrome. Through analogy with yeast, ALG14 is thought to form a multiglycosyltransferase complex with ALG13 and DPAGT1 that catalyses the first two committed steps of asparagine-linked protein glycosylation. We show that ALG14 is concentrated at the muscle motor endplates and small interfering RNA silencing of ALG14 results in reduced cell-surface expression of muscle acetylcholine receptor expressed in human embryonic kidney 293 cells. ALG2 is an alpha-1,3-mannosyltransferase that also catalyses early steps in the asparagine-linked glycosylation pathway. Mutations were identified in two kinships, with mutation ALG2p.Val68Gly found to severely reduce ALG2 expression both in patient muscle, and in cell cultures. Identification of DPAGT1, ALG14 and ALG2 mutations as a cause of congenital myasthenic syndrome underscores the importance of asparagine-linked protein glycosylation for proper functioning of the neuromuscular junction. These syndromes form part of the wider spectrum of congenital disorders of glycosylation caused by impaired asparagine-linked glycosylation. It is likely that further genes encoding components of this pathway will be associated with congenital myasthenic syndromes or impaired neuromuscular transmission as part of a more severe multisystem disorder. Our findings suggest that treatment with cholinesterase inhibitors may improve muscle function in many of the congenital disorders of glycosylation.
Figures

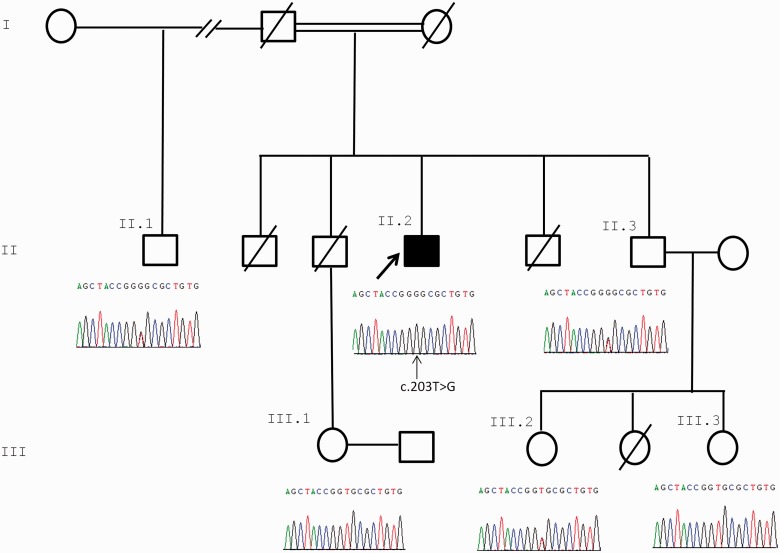
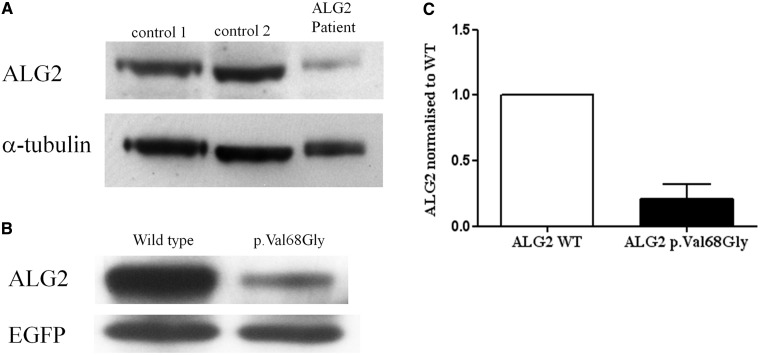
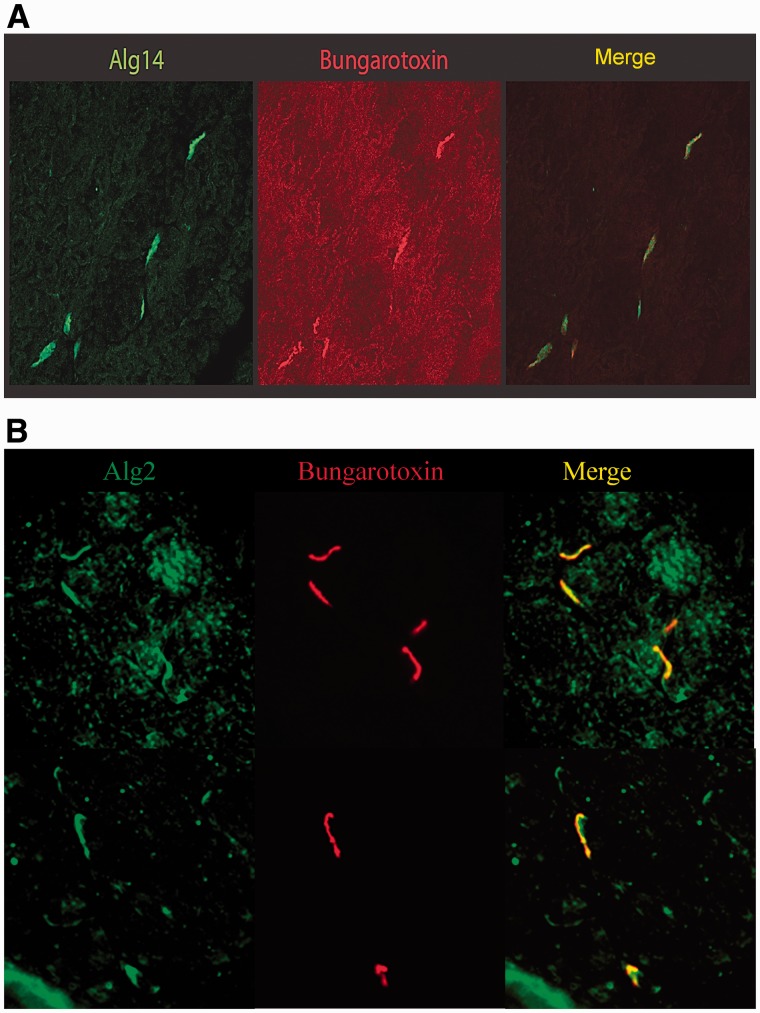
Comment in
-
Defective N-linked protein glycosylation pathway in congenital myasthenic syndromes.Brain. 2013 Mar;136(Pt 3):692-5. doi: 10.1093/brain/awt042. Brain. 2013. PMID: 23436500 Free PMC article. No abstract available.
References
-
- Beitz E. T(E)Xtopo: shaded membrane protein topology plots in LAT(E)X2epsilon. Bioinformatics. 2000;16:1050–1. - PubMed
-
- Bretthauer RK. Structure, expression, and regulation of UDP-GlcNAc: dolichol phosphate GlcNAc-1-phosphate transferase (DPAGT1) Curr Drug Targets. 2009;10:477–82. - PubMed
-
- Chaouch A, Beeson D, Hantai D, Lochmuller H. 186th ENMC international workshop: congenital myasthenic syndromes 24–26 June 2011, Naarden, The Netherlands. Neuromuscul Disord. 2012;22:566–76. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
