

A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis
- PMID: 23404504
- PMCID: PMC3611014
- DOI: 10.1074/jbc.M112.438010
A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis
Abstract
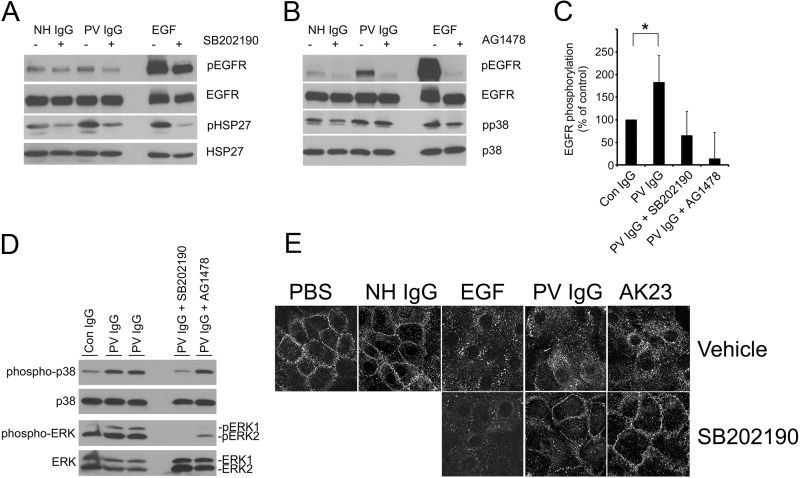
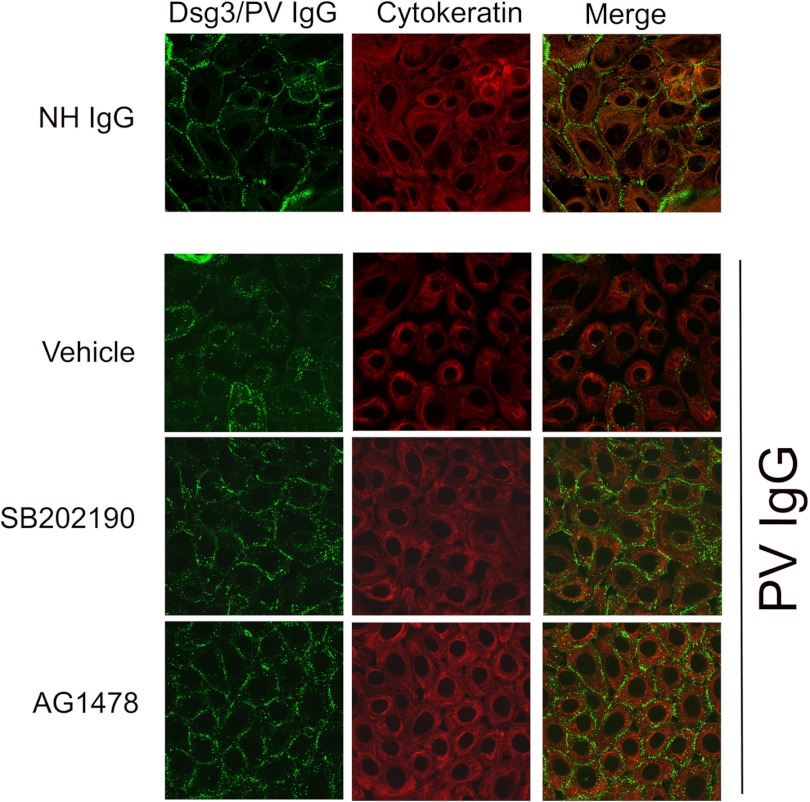
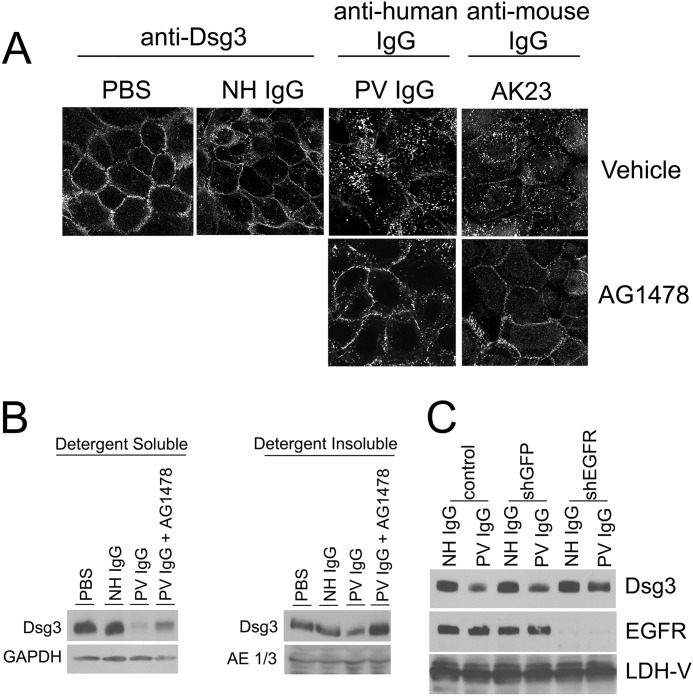
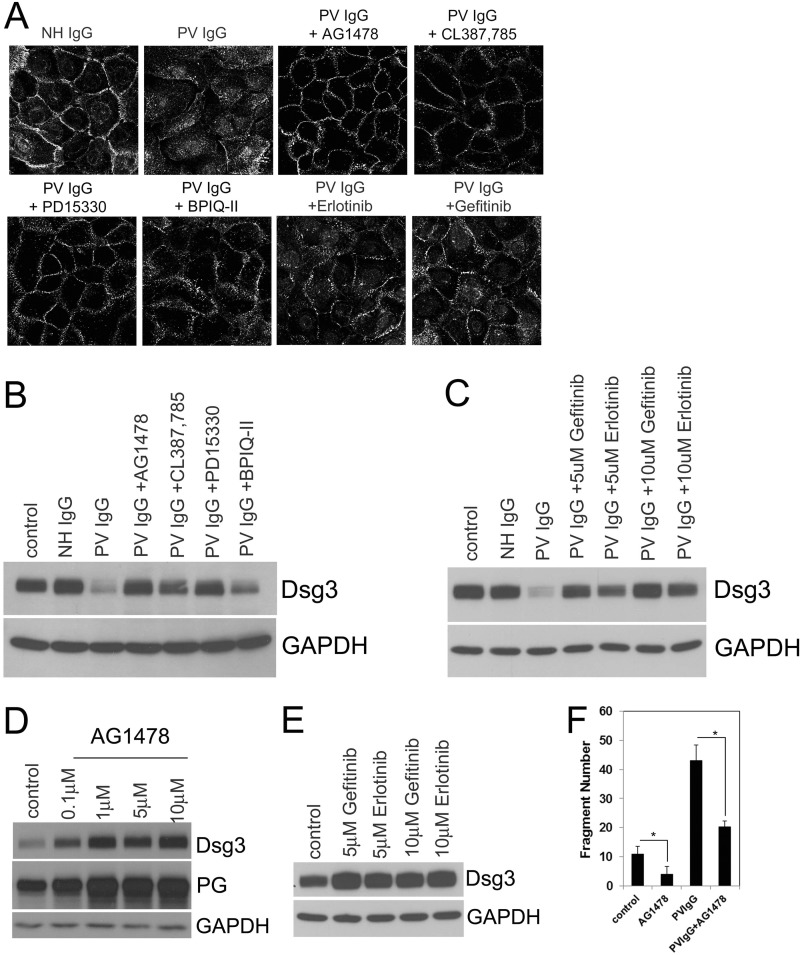
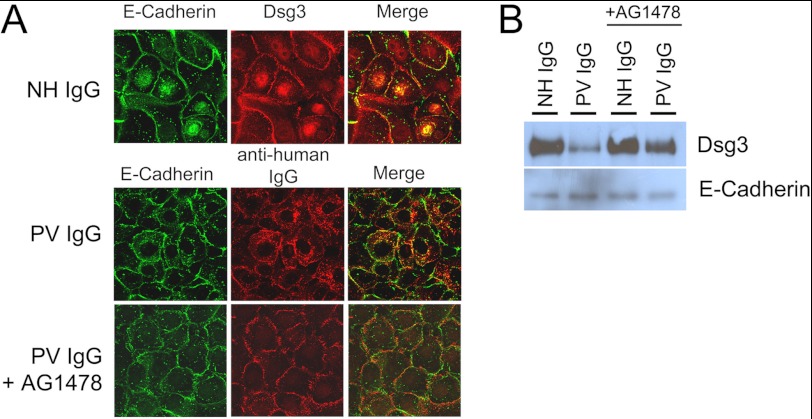
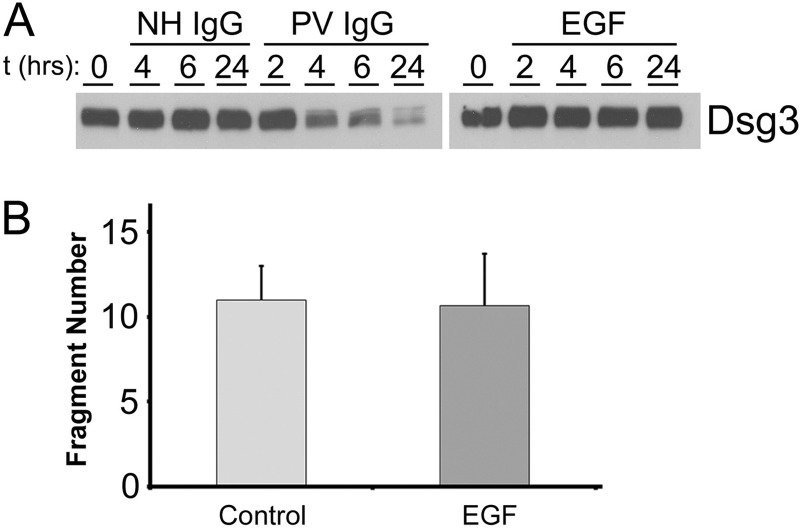
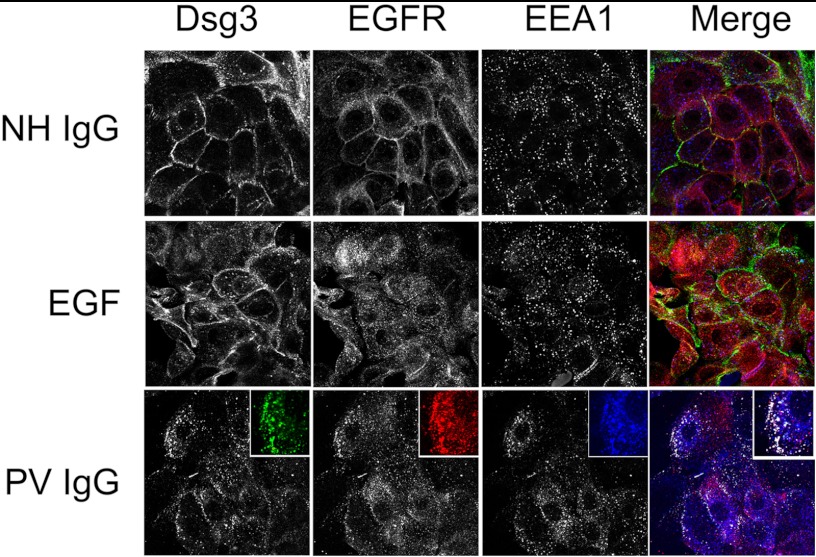
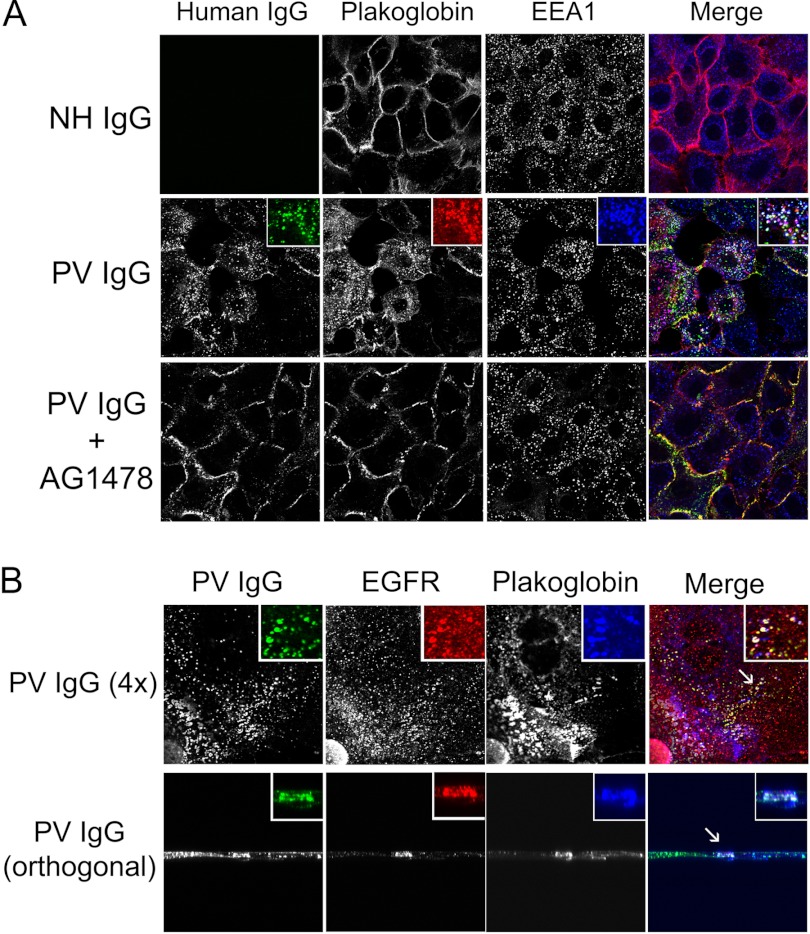
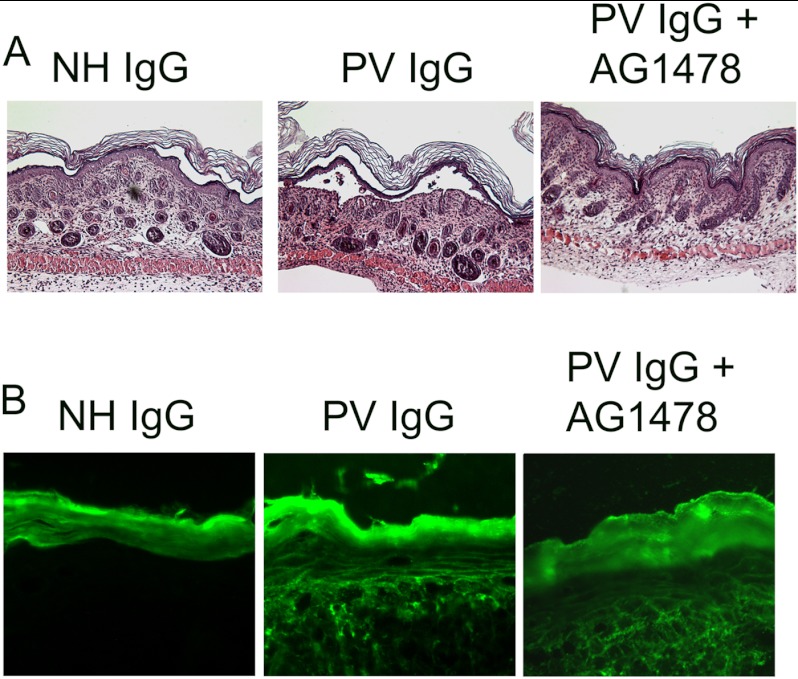
The pemphigus family of autoimmune bullous disorders is characterized by autoantibody binding to desmoglein 1 and/or 3 (dsg1/dsg3). In this study we show that EGF receptor (EGFR) is activated following pemphigus vulgaris (PV) IgG treatment of primary human keratinocytes and that EGFR activation is downstream of p38 mitogen-activated protein kinase (p38). Inhibition of EGFR blocked PV IgG-triggered dsg3 endocytosis, keratin intermediate filament retraction, and loss of cell-cell adhesion in vitro. Significantly, inhibiting EGFR prevented PV IgG-induced blister formation in the passive transfer mouse model of pemphigus. These data demonstrate cross-talk between dsg3 and EGFR, that this cross-talk is regulated by p38, and that EGFR is a potential therapeutic target for pemphigus. Small-molecule inhibitors and monoclonal antibodies directed against EGFR are currently used to treat several types of solid tumors. This study provides the experimental rationale for investigating the use of EGFR inhibitors in pemphigus.
Figures
References
-
- Stanley J. R., Koulu L., Klaus-Kovtun V., Steinberg M. S. (1986) A monoclonal antibody to the desmosomal glycoprotein desmoglein I binds the same polypeptide as human autoantibodies in pemphigus foliaceus. J. Immunol. 136, 1227–1230 - PubMed
-
- Amagai M., Klaus-Kovtun V., Stanley J. R. (1991) Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67, 869–877 - PubMed
-
- Seishima M., Iwasaki-Bessho Y., Itoh Y., Nozawa Y., Amagai M., Kitajima Y. (1999) Phosphatidylcholine-specific phospholipase C, but not phospholipase D, is involved in pemphigus IgG-induced signal transduction. Arch. Dermatol. Res. 291, 606–613 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
