

α₁A-adrenergic receptors regulate cardiac hypertrophy in vivo through interleukin-6 secretion
- PMID: 23404509
- PMCID: PMC3629827
- DOI: 10.1124/mol.112.084483
α₁A-adrenergic receptors regulate cardiac hypertrophy in vivo through interleukin-6 secretion
Abstract
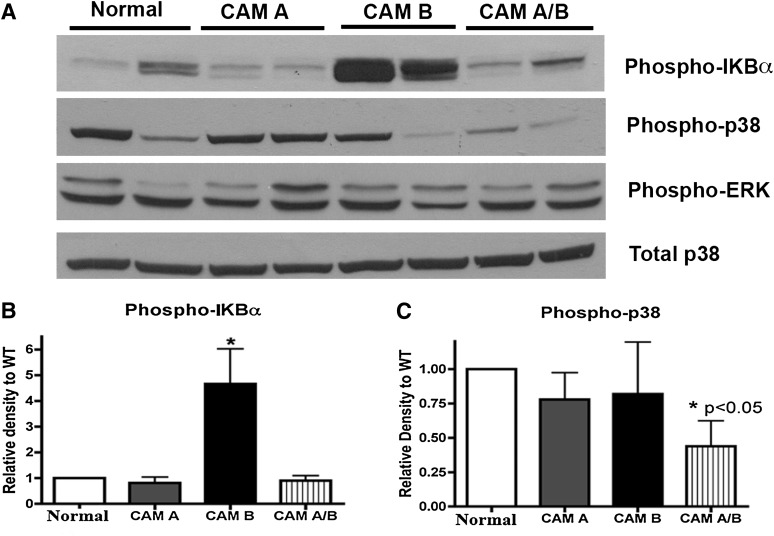
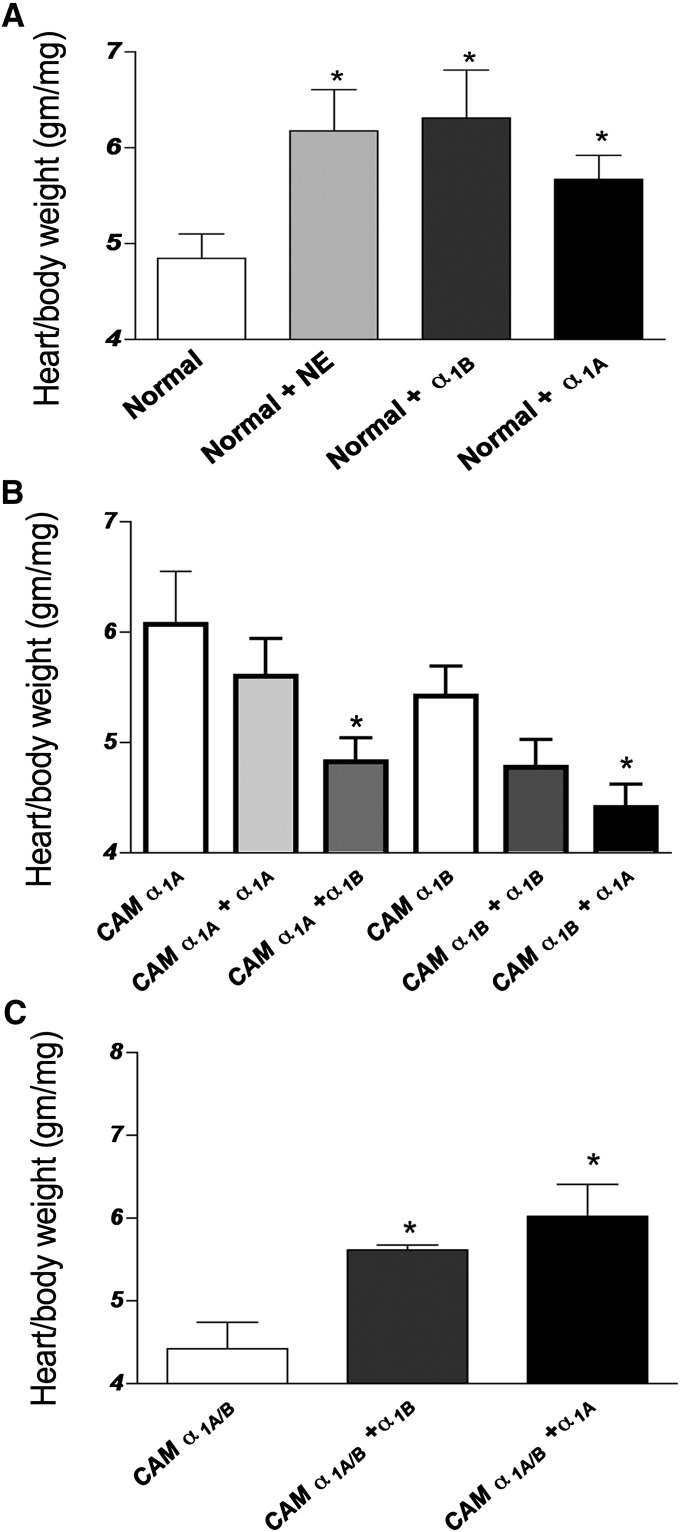
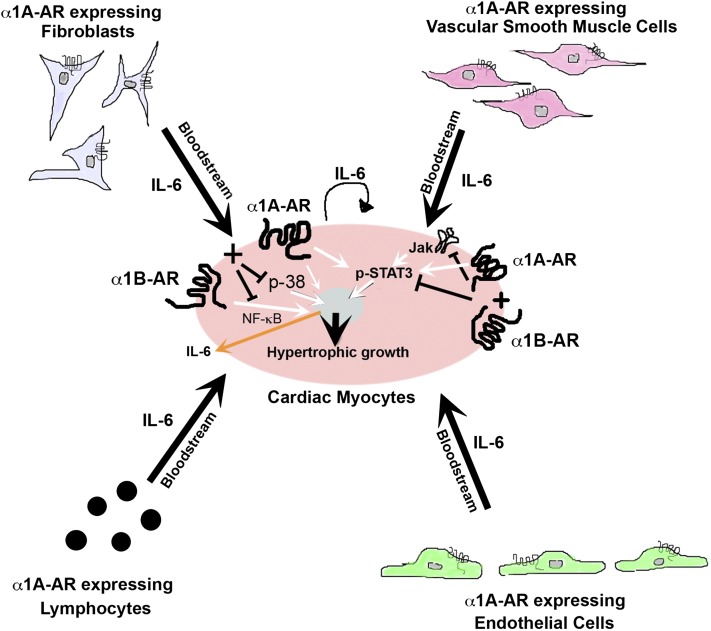
The role of α₁-adrenergic receptors (ARs) in the regulation of cardiac hypertrophy is still unclear, because transgenic mice demonstrated hypertrophy or the lack of it despite high receptor overexpression. To further address the role of the α₁-ARs in cardiac hypertrophy, we analyzed unique transgenic mice that overexpress constitutively active mutation (CAM) α₁A-ARs or CAM α₁B-ARs under the regulation of large fragments of their native promoters. These constitutively active receptors are expressed in all tissues that endogenously express their wild-type counterparts as opposed to only myocyte-targeted transgenic mice. In this study, we discovered that CAM α₁A-AR mice in vivo have cardiac hypertrophy independent of changes in blood pressure, corroborating earlier studies, but in contrast to myocyte-targeted α₁A-AR mice. We also found cardiac hypertrophy in CAM α₁B-AR mice, in agreement with previous studies, but hypertrophy only developed in older mice. We also discovered unique α₁-AR-mediated hypertrophic signaling that was AR subtype-specific with CAM α₁A-AR mice secreting atrial naturietic factor and interleukin-6 (IL-6), whereas CAM α₁B-AR mice expressed activated nuclear factor-κB (NF-κB). These particular hypertrophic signals were blocked when the other AR subtype was coactivated. We also discovered that crossbreeding the two CAM models (double CAM α₁A/B-AR) inhibited the development of hypertrophy and was reversible with single receptor activation, suggesting that coactivation of the receptors can lead to novel antagonistic signal transduction. This was confirmed by demonstrating antagonistic signals that were even lower than normal controls in the double CAM α₁A/B-AR mice for p38, NF-κB, and the IL-6/glycoprotein 130/signal transducer and activator of transcription 3 pathway. Because α₁A/B double knockout mice fail to develop hypertrophy in response to IL-6, our results suggest that IL-6 is a major mediator of α₁A-AR cardiac hypertrophy.
Figures





Similar articles
-
alpha1-Adrenergic receptor stimulates interleukin-6 expression and secretion through both mRNA stability and transcriptional regulation: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB.Mol Pharmacol. 2009 Jul;76(1):144-52. doi: 10.1124/mol.108.054320. Epub 2009 Apr 10. Mol Pharmacol. 2009. PMID: 19363165 Free PMC article.
-
Hybrid transgenic mice reveal in vivo specificity of G protein-coupled receptor kinases in the heart.Circ Res. 2000 Jan 7-21;86(1):43-50. doi: 10.1161/01.res.86.1.43. Circ Res. 2000. PMID: 10625304
-
Both alpha(1A)- and alpha(1B)-adrenergic receptors crosstalk to down regulate beta(1)-ARs in mouse heart: coupling to differential PTX-sensitive pathways.J Mol Cell Cardiol. 2005 Nov;39(5):777-84. doi: 10.1016/j.yjmcc.2005.07.015. Epub 2005 Sep 19. J Mol Cell Cardiol. 2005. PMID: 16171811
-
The alpha1-adrenergic receptors in cardiac hypertrophy: signaling mechanisms and functional implications.Cell Signal. 2015 Oct;27(10):1984-93. doi: 10.1016/j.cellsig.2015.06.009. Epub 2015 Jul 10. Cell Signal. 2015. PMID: 26169957 Review.
-
Adrenergic regulation of cardiac myocyte apoptosis.J Cell Physiol. 2001 Dec;189(3):257-65. doi: 10.1002/jcp.10024. J Cell Physiol. 2001. PMID: 11748583 Review.
Cited by
-
Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance.Pharmacol Rev. 2013 Dec 24;66(1):308-33. doi: 10.1124/pr.112.007203. Print 2014. Pharmacol Rev. 2013. PMID: 24368739 Free PMC article. Review.
-
Current Developments on the Role of α1-Adrenergic Receptors in Cognition, Cardioprotection, and Metabolism.Front Cell Dev Biol. 2021 May 25;9:652152. doi: 10.3389/fcell.2021.652152. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34113612 Free PMC article. Review.
-
Angiotensin II-induced cardiovascular load regulates cardiac remodeling and related gene expression in late-gestation fetal sheep.Pediatr Res. 2014 Jun;75(6):689-696. doi: 10.1038/pr.2014.37. Epub 2014 Mar 10. Pediatr Res. 2014. PMID: 24614802 Free PMC article.
-
The Antiarrhythmic Activity of Novel Pyrrolidin-2-one Derivative S-75 in Adrenaline-Induced Arrhythmia.Pharmaceuticals (Basel). 2021 Oct 20;14(11):1065. doi: 10.3390/ph14111065. Pharmaceuticals (Basel). 2021. PMID: 34832847 Free PMC article.
-
Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO.Front Physiol. 2018 Apr 11;9:382. doi: 10.3389/fphys.2018.00382. eCollection 2018. Front Physiol. 2018. PMID: 29695980 Free PMC article. Review.
References
-
- Autelitano DJ, Woodcock EA. (1998) Selective activation of α1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of α1-adrenergic receptor subtype mRNAs. J Mol Cell Cardiol 30:1515–1523 - PubMed
-
- Butler KL, Huffman LC, Koch SE, Hahn HS, Gwathmey JK. (2006) STAT-3 activation is necessary for ischemic preconditioning in hypertrophied myocardium. Am J Physiol Heart Circ Physiol 291:H797–H803 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
