

Antibiotics as selectors and accelerators of diversity in the mechanisms of resistance: from the resistome to genetic plasticity in the β-lactamases world
- PMID: 23404545
- PMCID: PMC3567504
- DOI: 10.3389/fmicb.2013.00009
Antibiotics as selectors and accelerators of diversity in the mechanisms of resistance: from the resistome to genetic plasticity in the β-lactamases world
Abstract
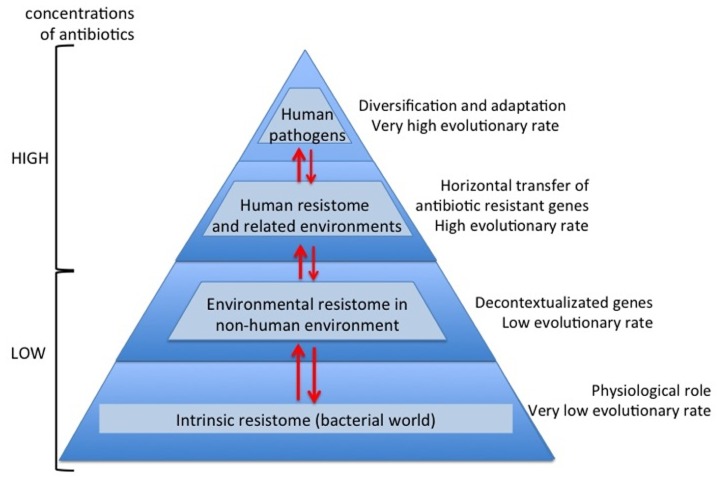
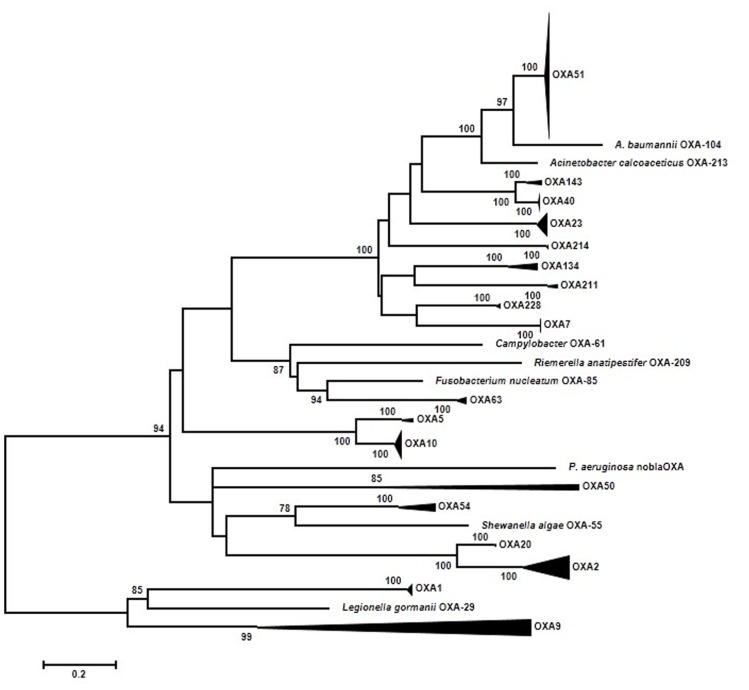
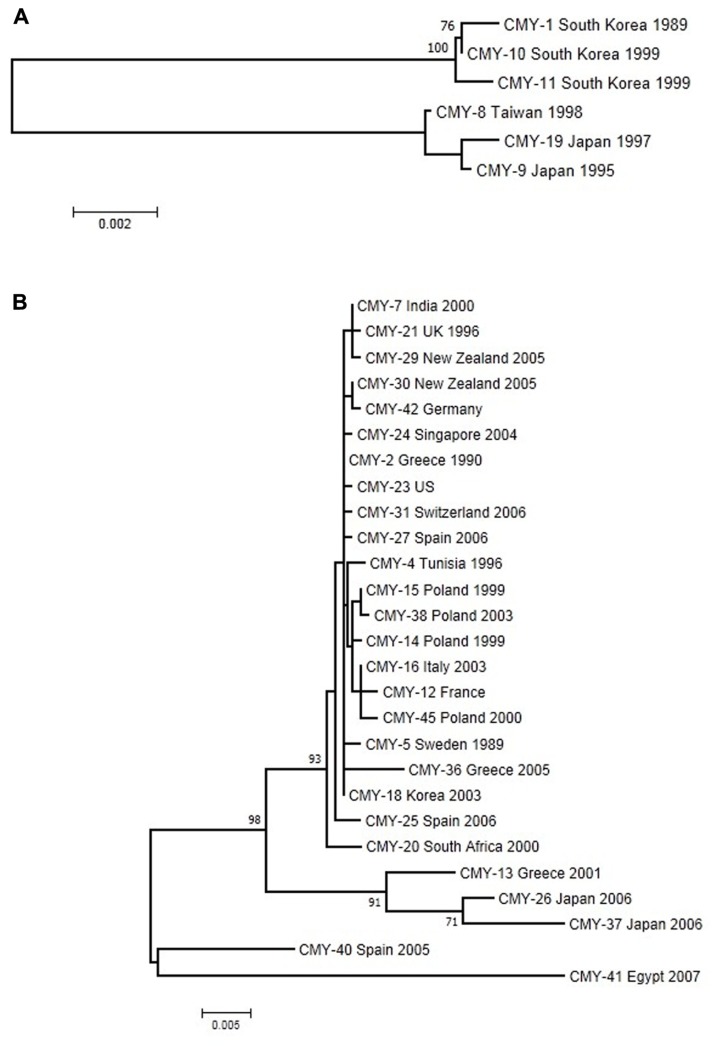
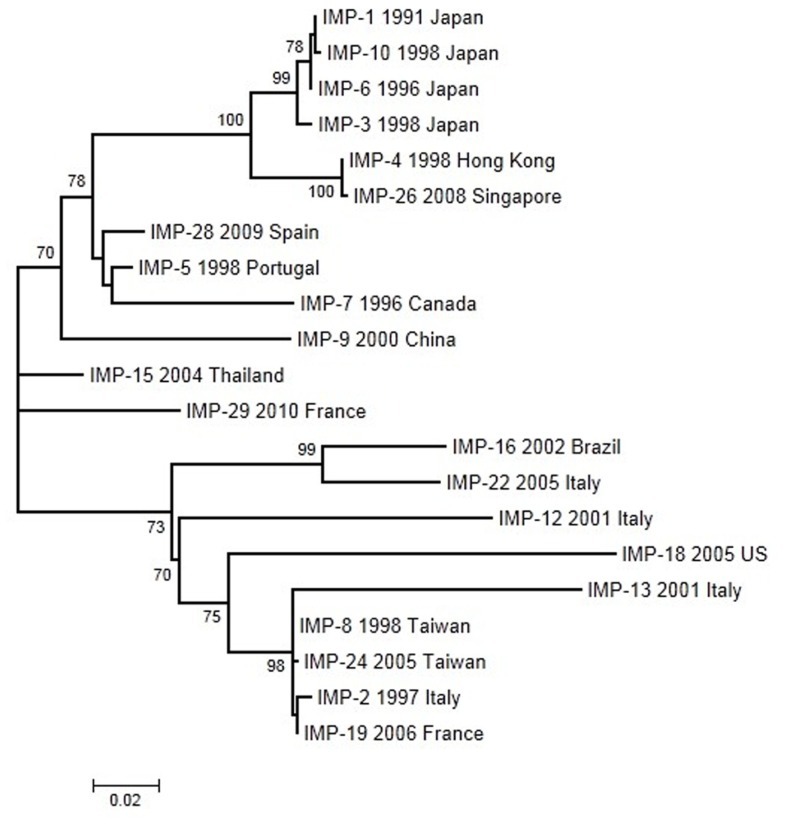
Antibiotics and antibiotic resistance determinants, natural molecules closely related to bacterial physiology and consistent with an ancient origin, are not only present in antibiotic-producing bacteria. Throughput sequencing technologies have revealed an unexpected reservoir of antibiotic resistance in the environment. These data suggest that co-evolution between antibiotic and antibiotic resistance genes has occurred since the beginning of time. This evolutionary race has probably been slow because of highly regulated processes and low antibiotic concentrations. Therefore to understand this global problem, a new variable must be introduced, that the antibiotic resistance is a natural event, inherent to life. However, the industrial production of natural and synthetic antibiotics has dramatically accelerated this race, selecting some of the many resistance genes present in nature and contributing to their diversification. One of the best models available to understand the biological impact of selection and diversification are β-lactamases. They constitute the most widespread mechanism of resistance, at least among pathogenic bacteria, with more than 1000 enzymes identified in the literature. In the last years, there has been growing concern about the description, spread, and diversification of β-lactamases with carbapenemase activity and AmpC-type in plasmids. Phylogenies of these enzymes help the understanding of the evolutionary forces driving their selection. Moreover, understanding the adaptive potential of β-lactamases contribute to exploration the evolutionary antagonists trajectories through the design of more efficient synthetic molecules. In this review, we attempt to analyze the antibiotic resistance problem from intrinsic and environmental resistomes to the adaptive potential of resistance genes and the driving forces involved in their diversification, in order to provide a global perspective of the resistance problem.
Keywords: environmental resistome; intrinsic resistome; plasticity; β-lactamase.
Figures
References
-
- Abriata L. A., Merijn L., Salverda M., Tomatis P. E. (2012). Sequence-function-stability relationships in proteins from datasets of functionally annotated variants: the case of TEM β-lactamases. FEBS Lett. 586 3330–3335 - PubMed
-
- Alekshun M. N., Levy S. B. (2007). Molecular mechanisms of antibacterial multidrug resistance. Cell 128 1037–1050 - PubMed
-
- Allen H. K., Donato J., Wang H. H., Cloud-Hansen K. A., Davies J., Handelsman J. (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8 251–259 - PubMed
-
- Allen H. K., Moe L. A., Rodbumrer J., Gaarder A., Handelsman J. (2009). Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J. 3 243–251 - PubMed
-
- Archetti M. (2009). Survival of the steepest: hypersensitivity to mutations as an adaptation to soft selection. J. Evol. Biol. 22 740–750 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources