

Direct activation of full-length proapoptotic BAK
- PMID: 23404709
- PMCID: PMC3600461
- DOI: 10.1073/pnas.1214313110
Direct activation of full-length proapoptotic BAK
Abstract
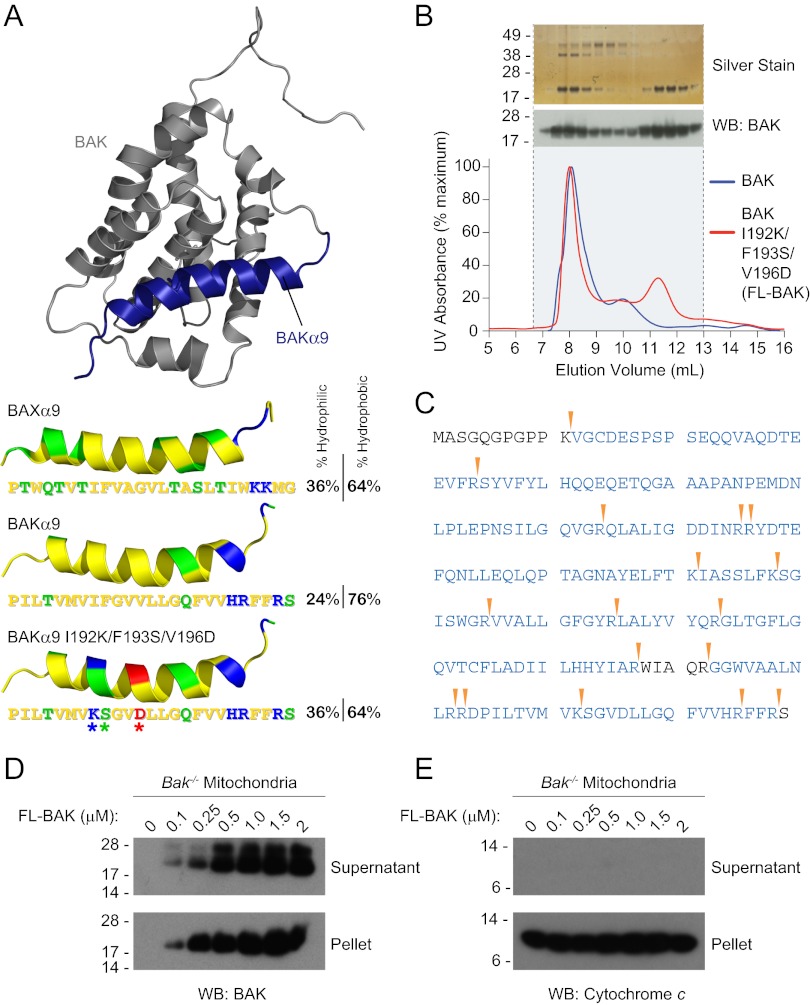
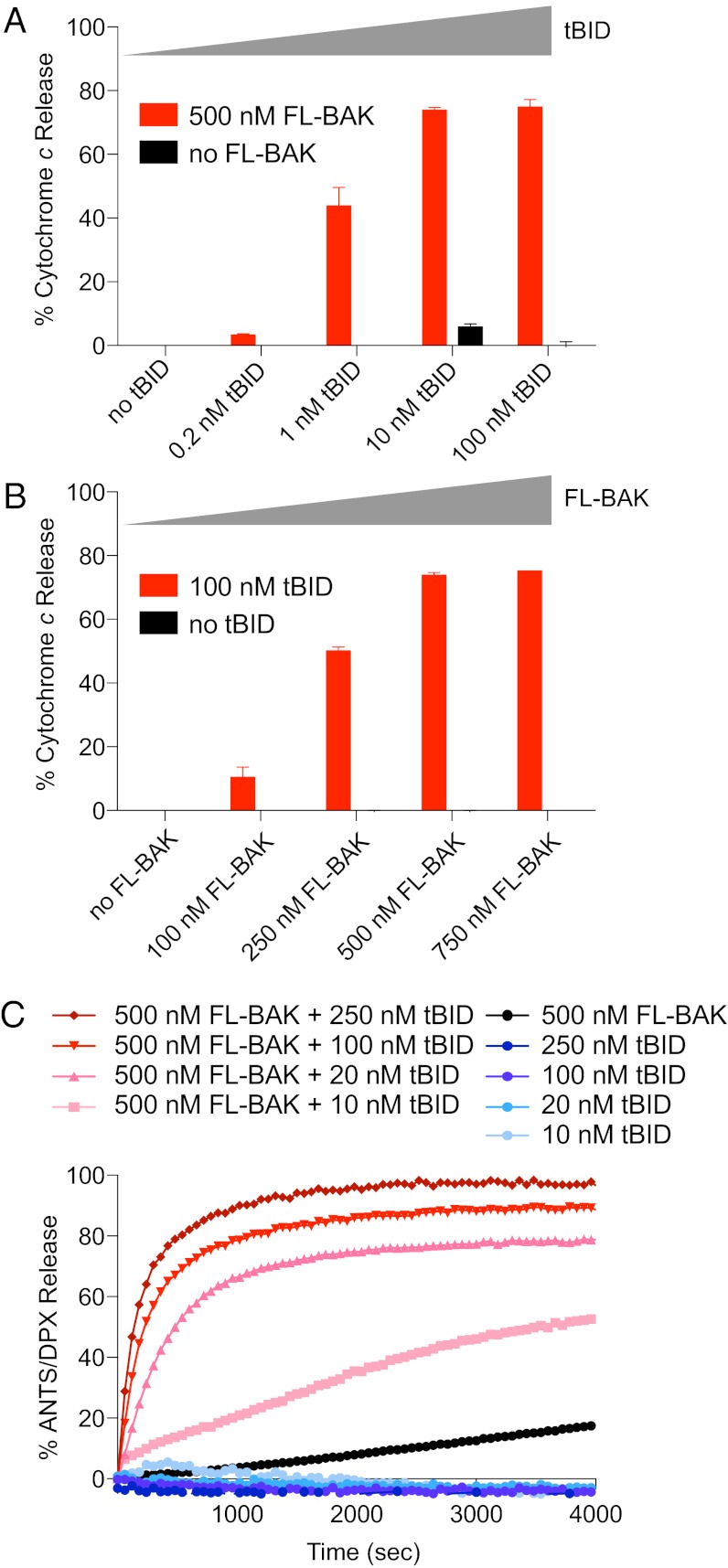
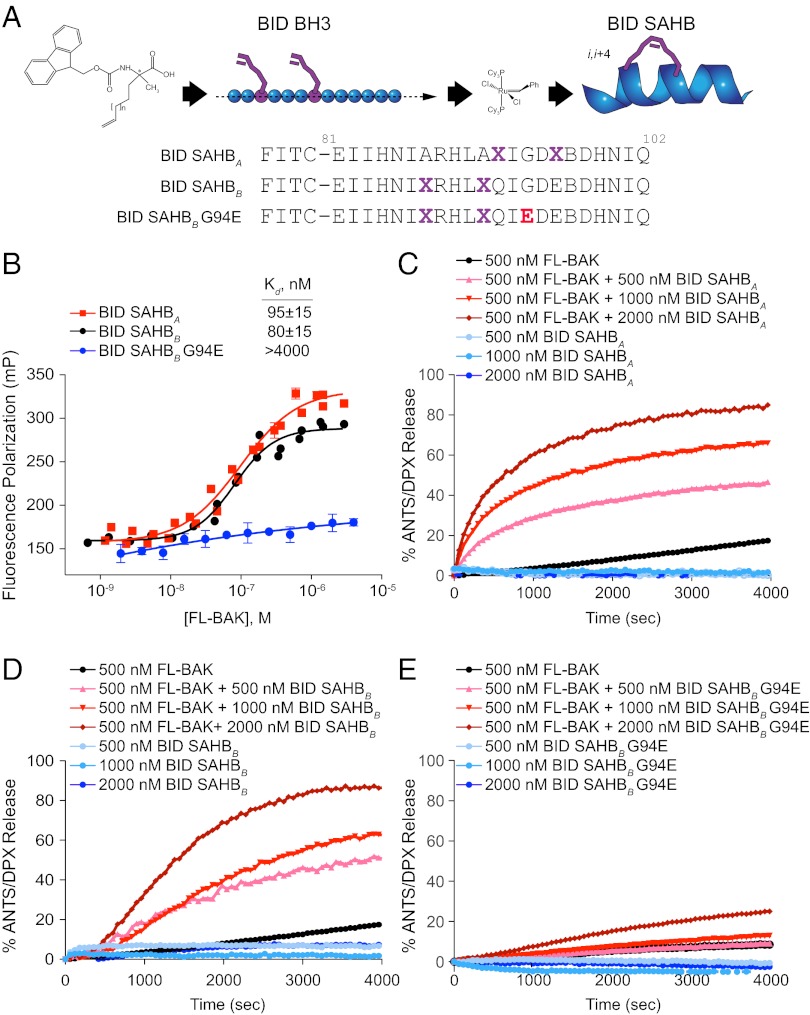
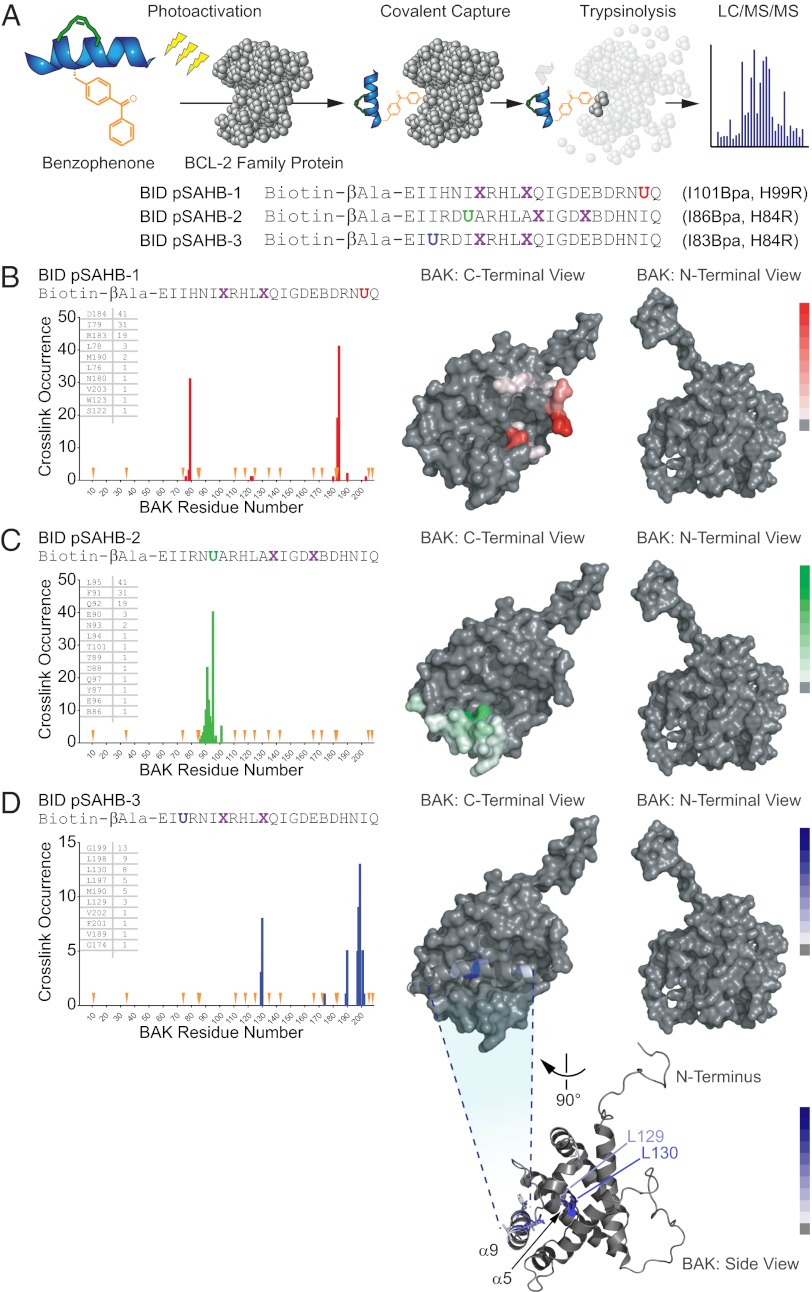
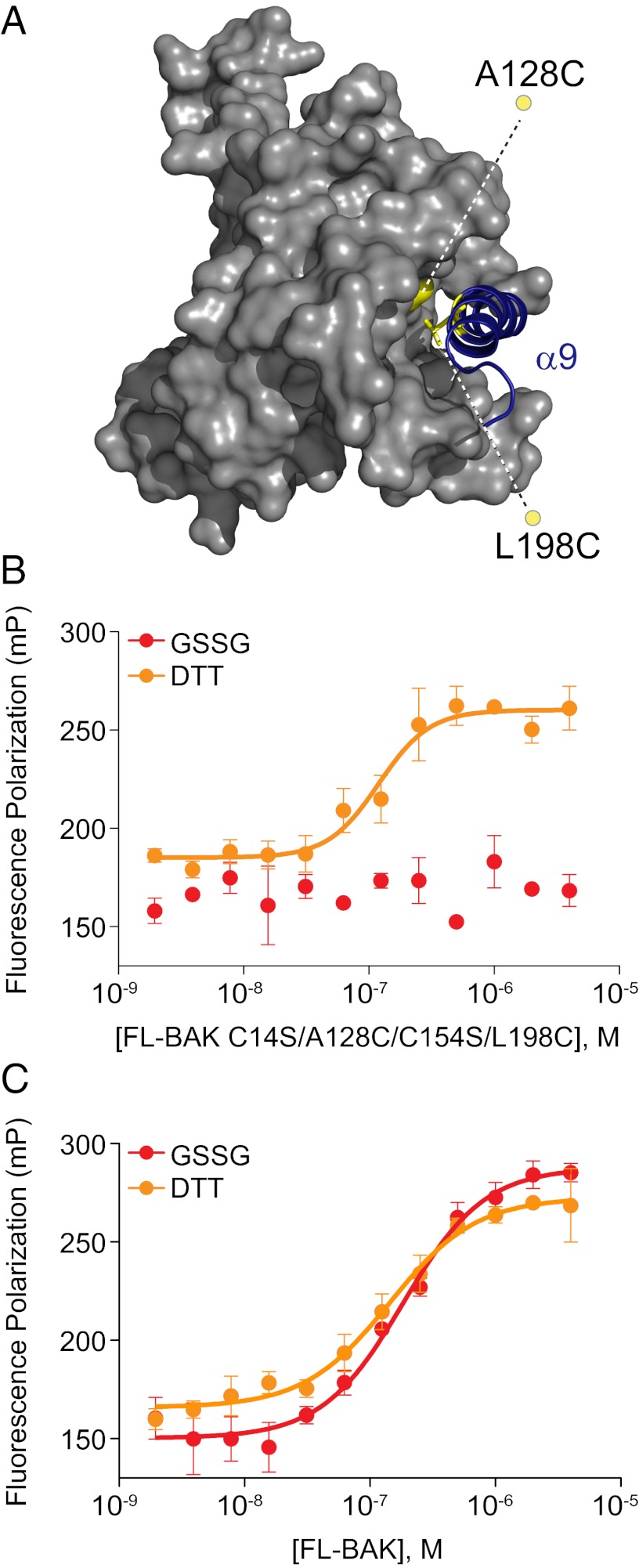
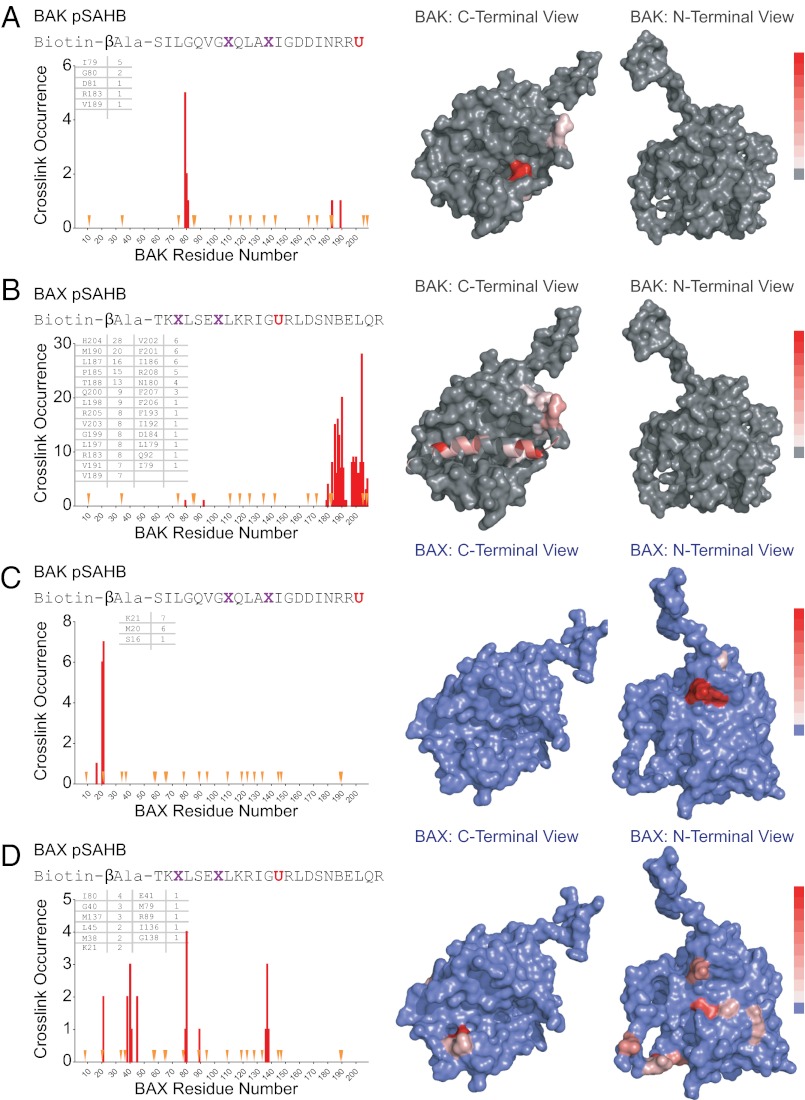
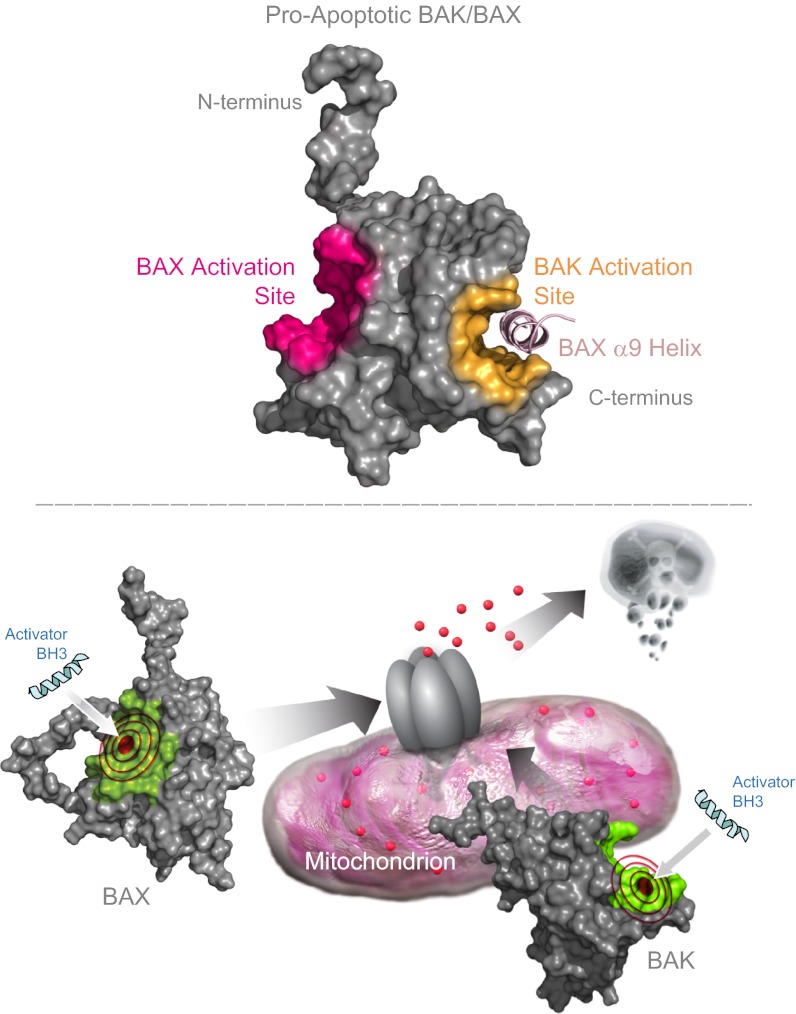
Proapoptotic B-cell lymphoma 2 (BCL-2) antagonist/killer (BAK) and BCL-2-associated X (BAX) form toxic mitochondrial pores in response to cellular stress. Whereas BAX resides predominantly in the cytosol, BAK is constitutively localized to the outer mitochondrial membrane. Select BCL-2 homology domain 3 (BH3) helices activate BAX directly by engaging an α1/α6 trigger site. The inability to express full-length BAK has hampered full dissection of its activation mechanism. Here, we report the production of full-length, monomeric BAK by mutagenesis-based solubilization of its C-terminal α-helical surface. Recombinant BAK autotranslocates to mitochondria but only releases cytochrome c upon BH3 triggering. A direct activation mechanism was explicitly demonstrated using a liposomal system that recapitulates BAK-mediated release upon addition of BH3 ligands. Photoreactive BH3 helices mapped both triggering and autointeractions to the canonical BH3-binding pocket of BAK, whereas the same ligands crosslinked to the α1/α6 site of BAX. Thus, activation of both BAK and BAX is initiated by direct BH3-interaction but at distinct trigger sites. These structural and biochemical insights provide opportunities for developing proapoptotic agents that activate the death pathway through direct but differential engagement of BAK and BAX.
Conflict of interest statement
Conflict of interest statement: L.D.W. is a scientific advisory board member and consultant for Aileron Therapeutics.
Figures
References
-
- Yang J, et al. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. - PubMed
-
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
