

GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit
- PMID: 23407358
- PMCID: PMC3605599
- DOI: 10.1093/bioinformatics/btt055
GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit
Abstract
Motivation: Molecular simulation has historically been a low-throughput technique, but faster computers and increasing amounts of genomic and structural data are changing this by enabling large-scale automated simulation of, for instance, many conformers or mutants of biomolecules with or without a range of ligands. At the same time, advances in performance and scaling now make it possible to model complex biomolecular interaction and function in a manner directly testable by experiment. These applications share a need for fast and efficient software that can be deployed on massive scale in clusters, web servers, distributed computing or cloud resources.
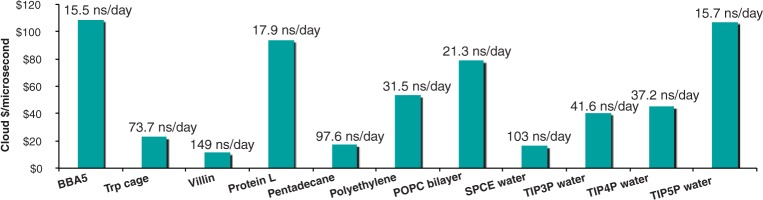
Results: Here, we present a range of new simulation algorithms and features developed during the past 4 years, leading up to the GROMACS 4.5 software package. The software now automatically handles wide classes of biomolecules, such as proteins, nucleic acids and lipids, and comes with all commonly used force fields for these molecules built-in. GROMACS supports several implicit solvent models, as well as new free-energy algorithms, and the software now uses multithreading for efficient parallelization even on low-end systems, including windows-based workstations. Together with hand-tuned assembly kernels and state-of-the-art parallelization, this provides extremely high performance and cost efficiency for high-throughput as well as massively parallel simulations.
Availability: GROMACS is an open source and free software available from http://www.gromacs.org.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
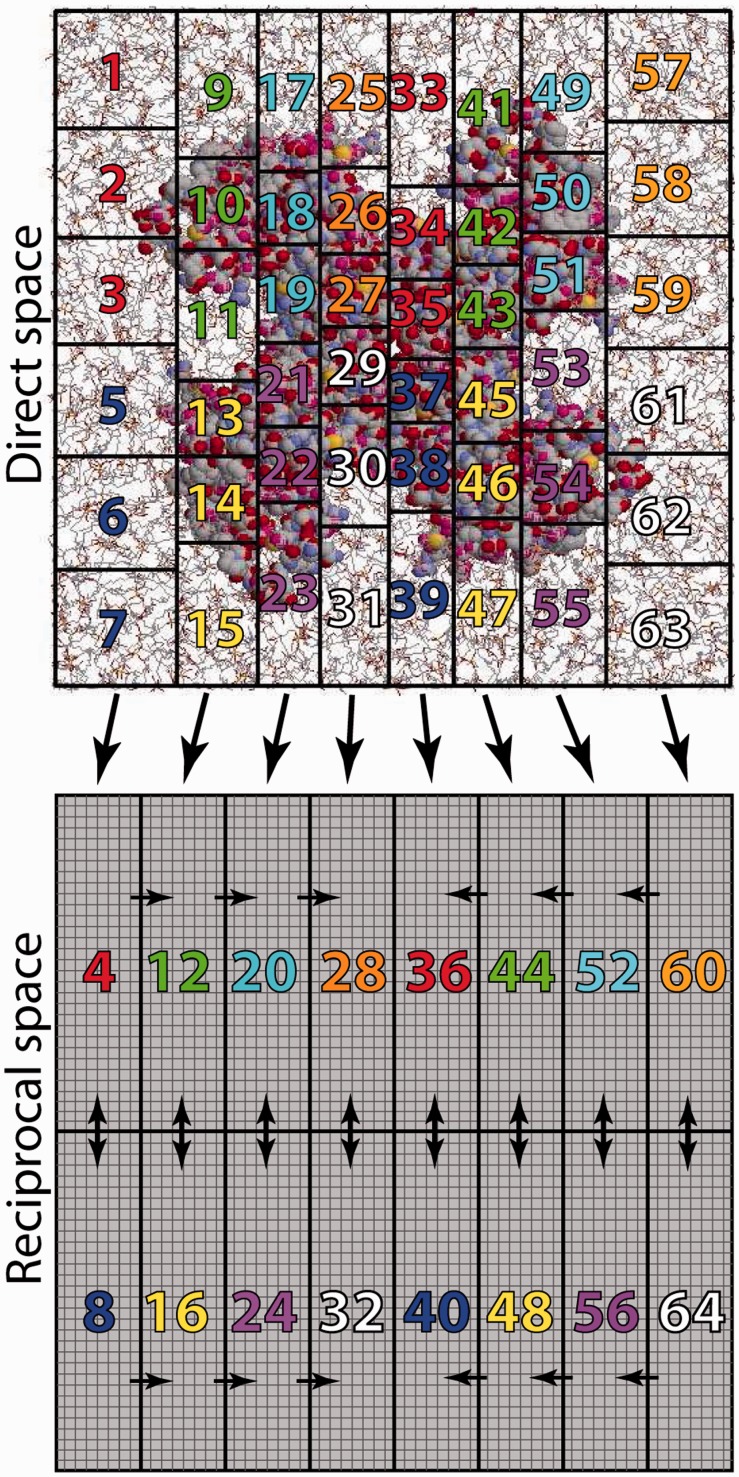
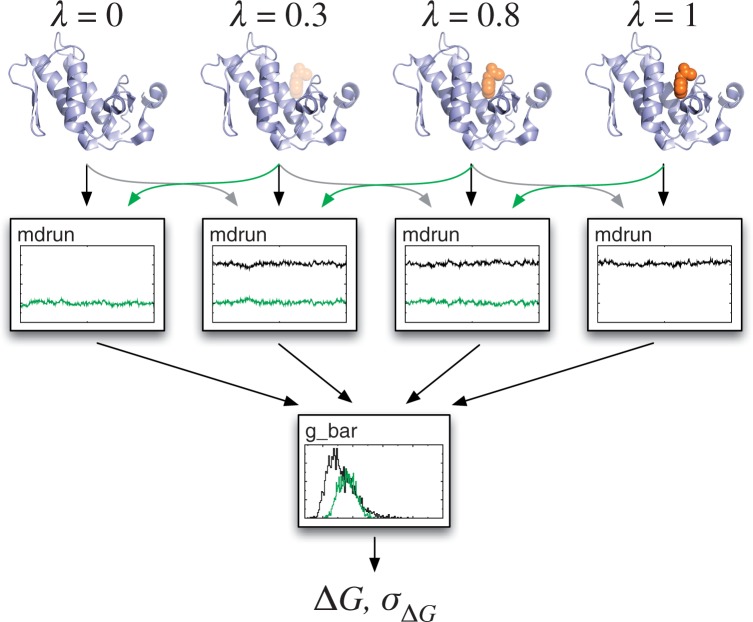
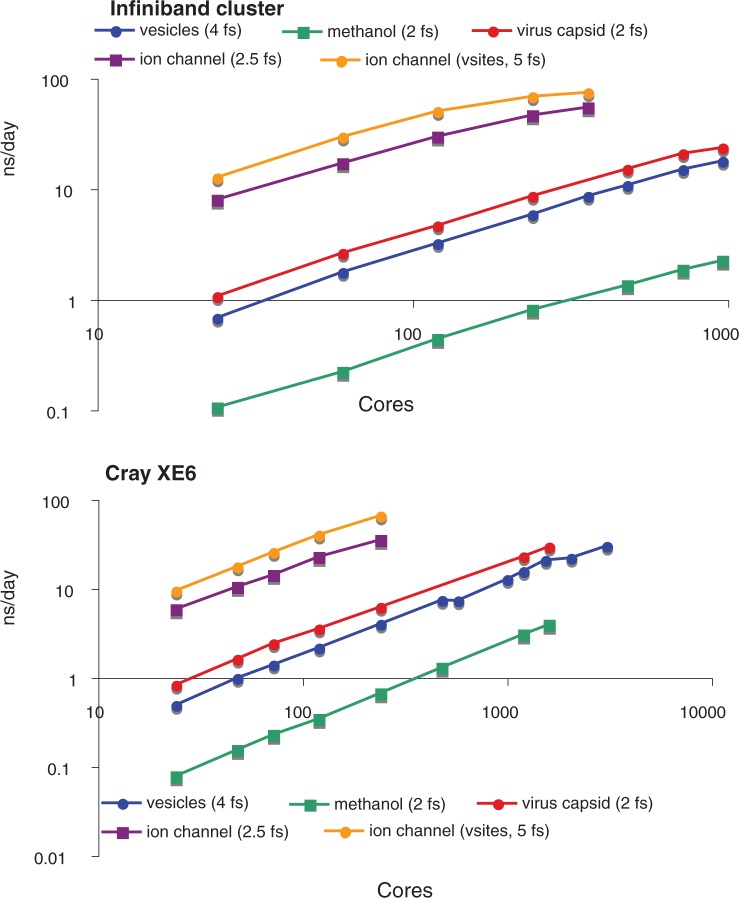
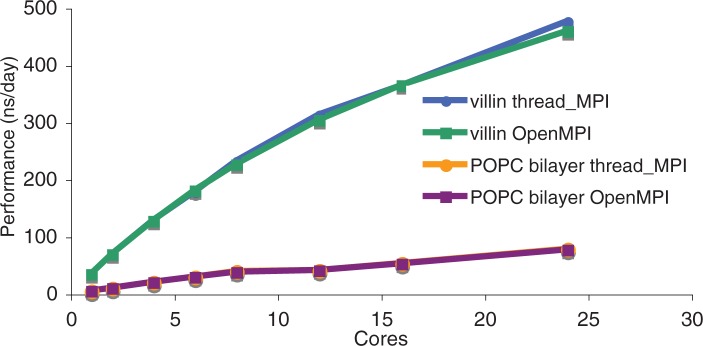
References
-
- Alder B, Wainwright T. Phase transition for a hard sphere system. J. Chem. Phys. 1957;27:1208–1209.
-
- Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. J. Comp. Phys. 1976;22:245.
-
- Berendsen H. Technical report. CECAM, Lyon: 1976. Report of CECAM workshop: models for protein dynamics.
-
- Bowers KJ, et al. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) ACM, New York, NY: 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
