

A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity
- PMID: 23407963
- PMCID: PMC3711767
- DOI: 10.1523/JNEUROSCI.3430-12.2013
A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity
Abstract
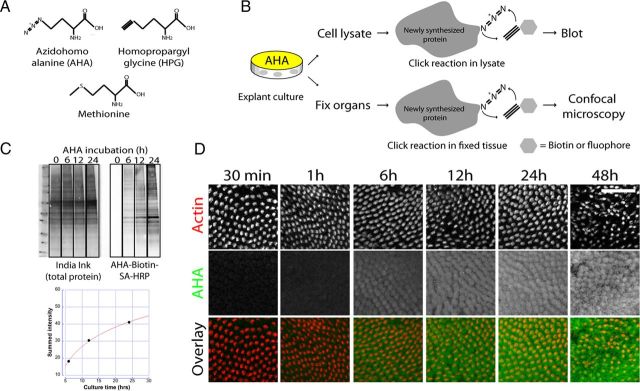
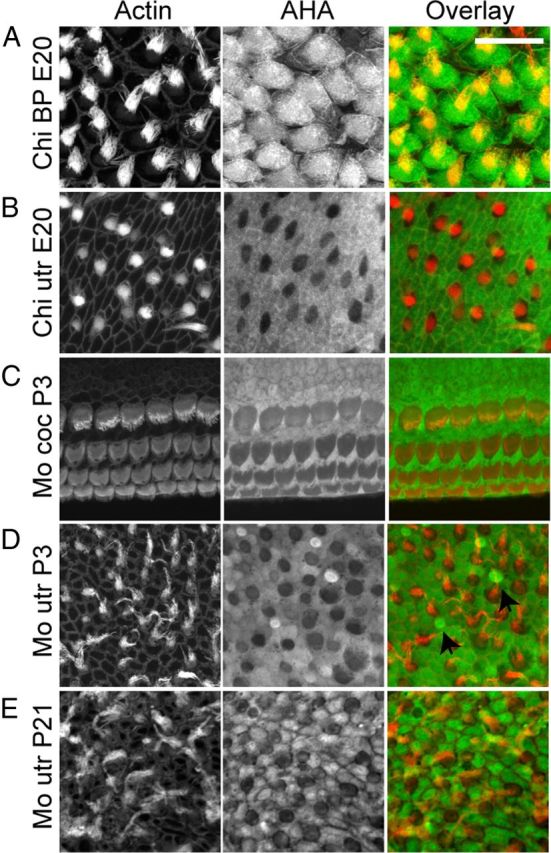
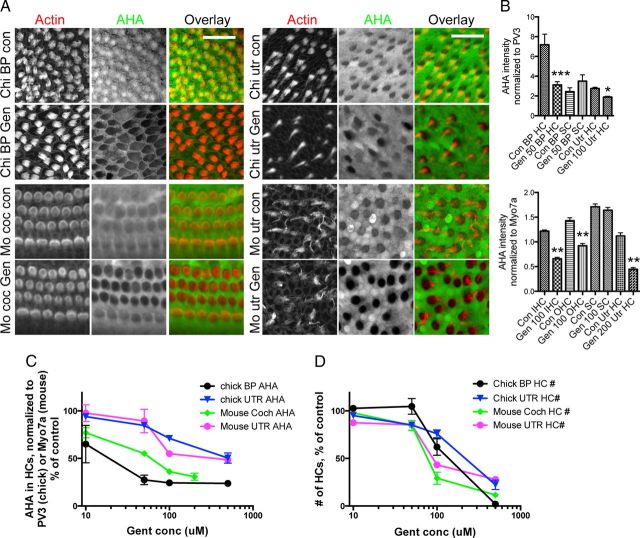
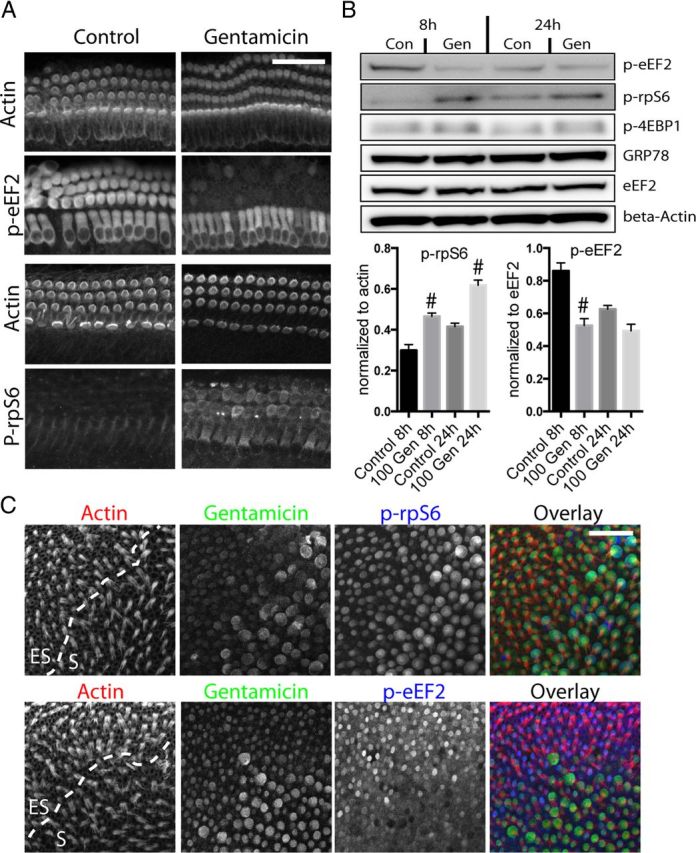
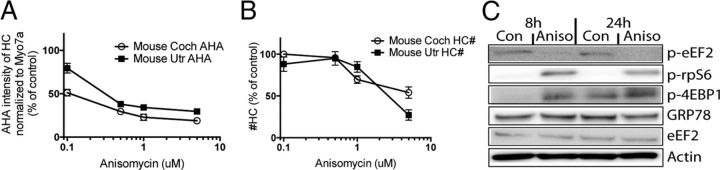
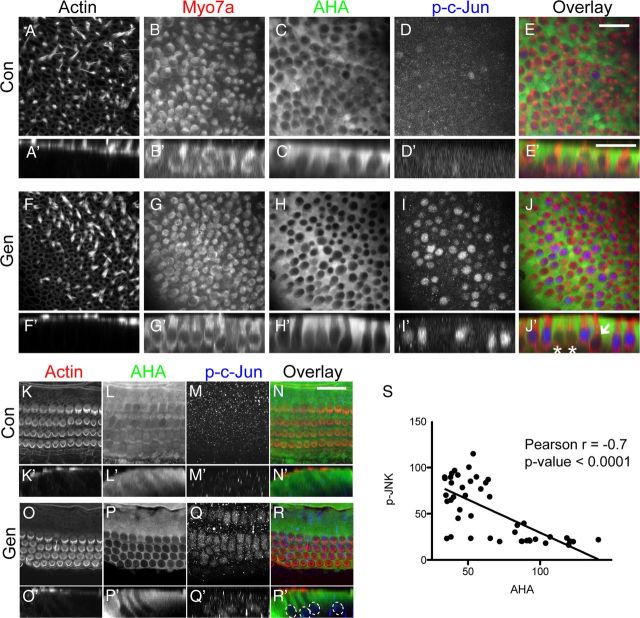
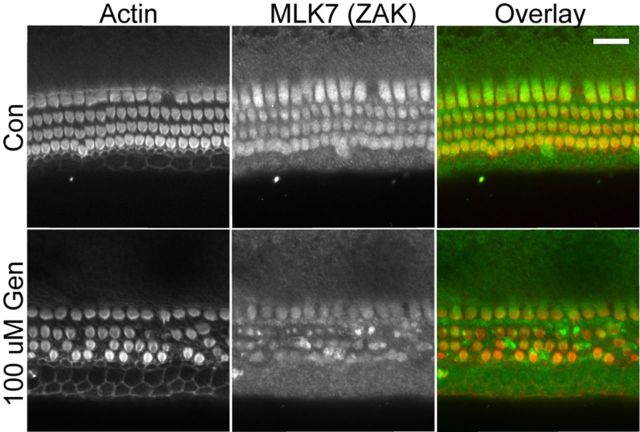
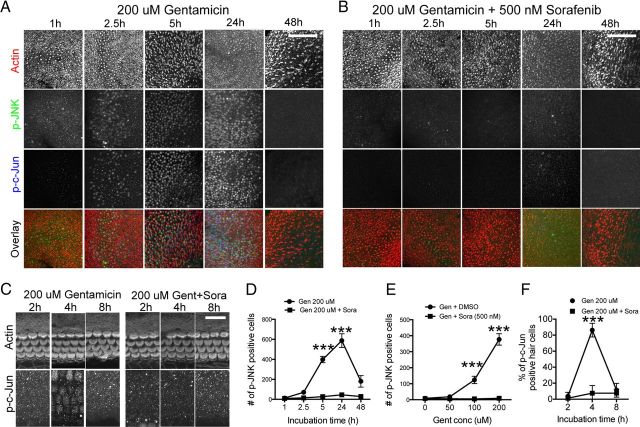
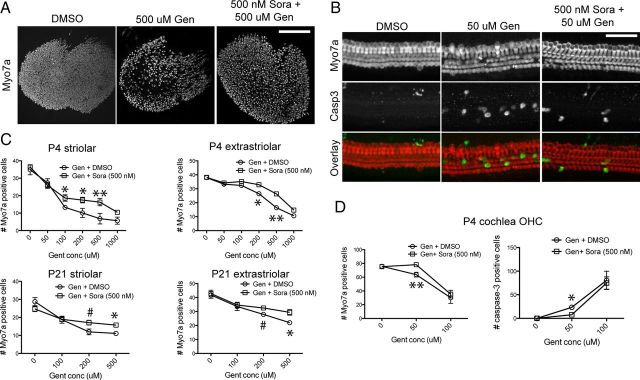
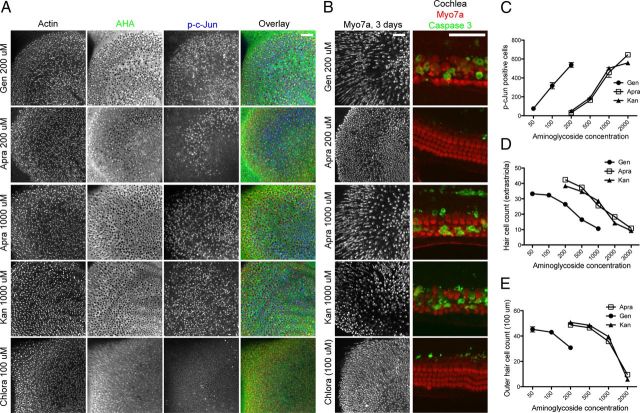
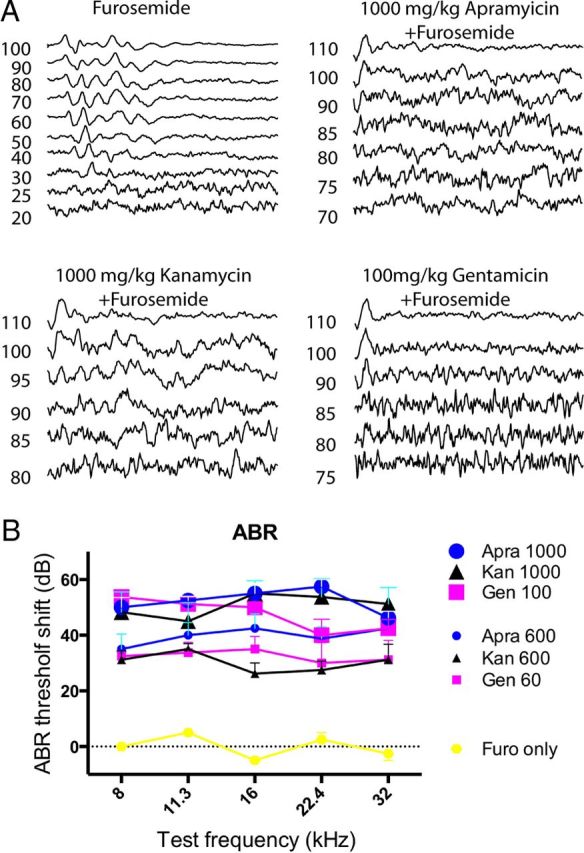
Ototoxicity is a main dose-limiting factor in the clinical application of aminoglycoside antibiotics. Despite longstanding research efforts, our understanding of the mechanisms underlying aminoglycoside ototoxicity remains limited. Here we report the discovery of a novel stress pathway that contributes to aminoglycoside-induced hair cell degeneration. Modifying the previously developed bioorthogonal noncanonical amino acid tagging method, we used click chemistry to study the role of protein synthesis activity in aminoglycoside-induced hair cell stress. We demonstrate that aminoglycosides inhibit protein synthesis in hair cells and activate a signaling pathway similar to ribotoxic stress response, contributing to hair cell degeneration. The ability of a particular aminoglycoside to inhibit protein synthesis and to activate the c-Jun N-terminal kinase (JNK) pathway correlated well with its ototoxic potential. Finally, we report that a Food and Drug Administration-approved drug known to inhibit ribotoxic stress response also prevents JNK activation and improves hair cell survival, opening up novel strategies to prevent and treat aminoglycoside ototoxicity.
Figures


References
-
- Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. - PubMed
-
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. - PubMed
-
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous