

Site- and strand-specific nicking of DNA by fusion proteins derived from MutH and I-SceI or TALE repeats
- PMID: 23408850
- PMCID: PMC3627573
- DOI: 10.1093/nar/gkt080
Site- and strand-specific nicking of DNA by fusion proteins derived from MutH and I-SceI or TALE repeats
Abstract
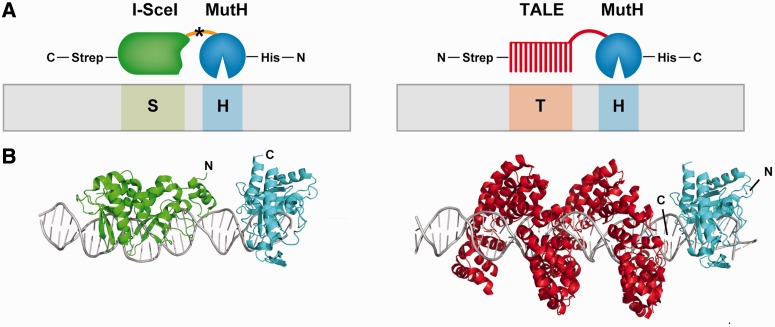
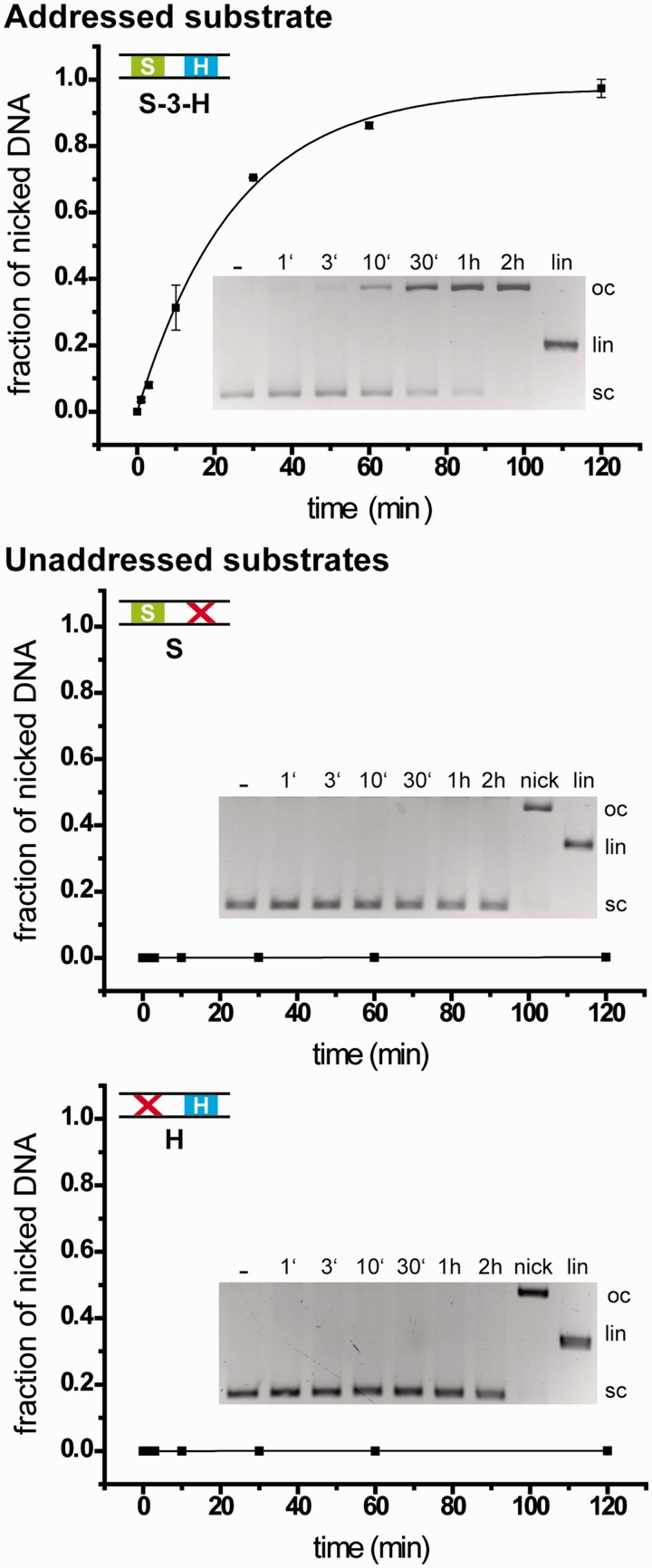
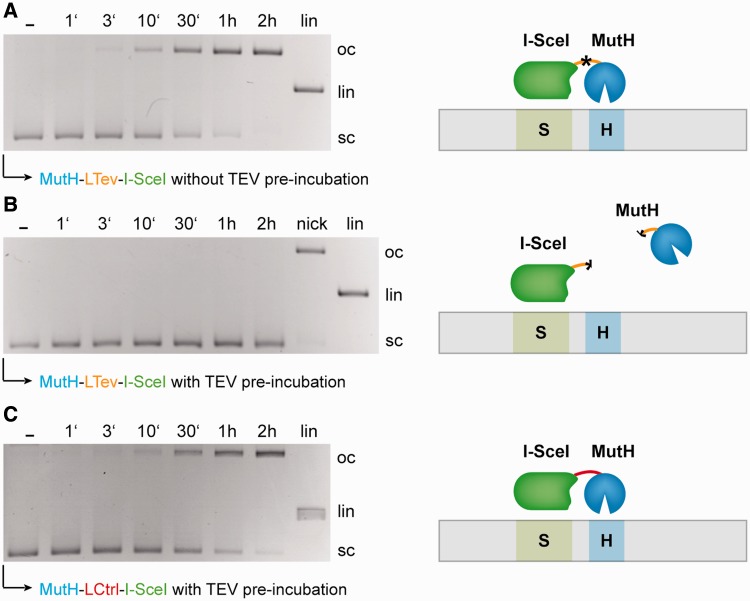
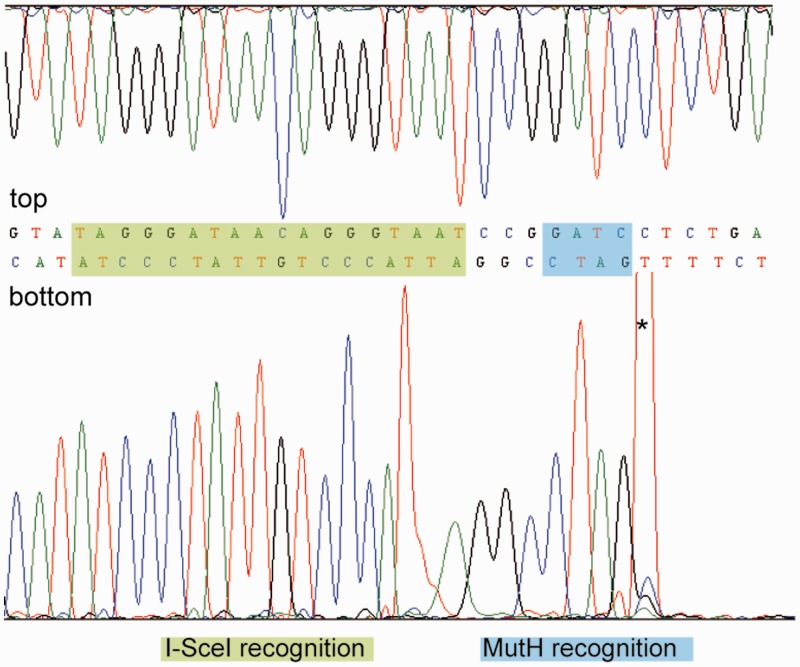
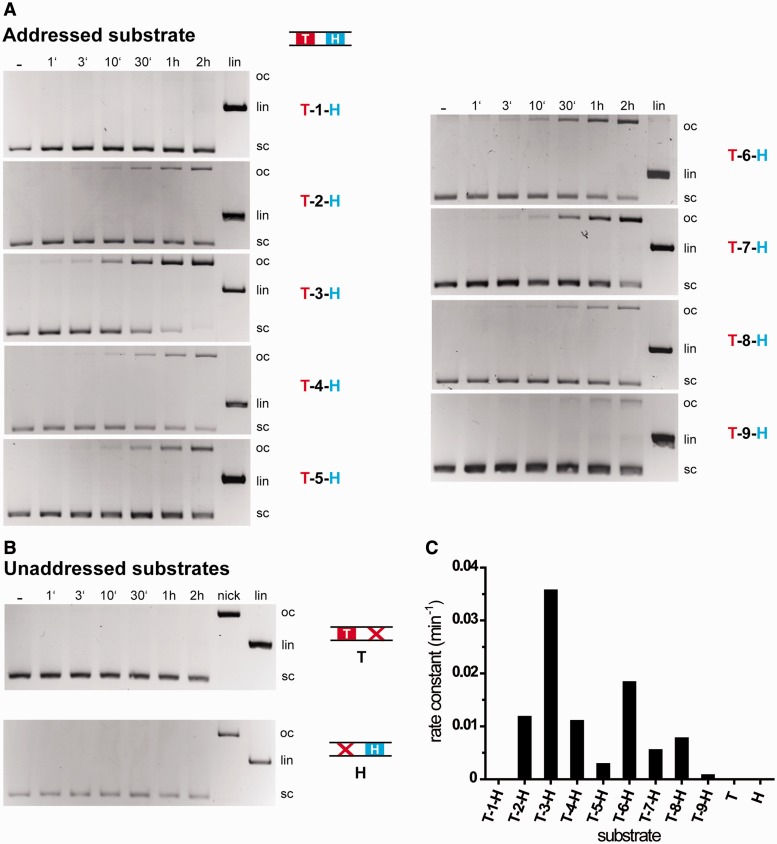
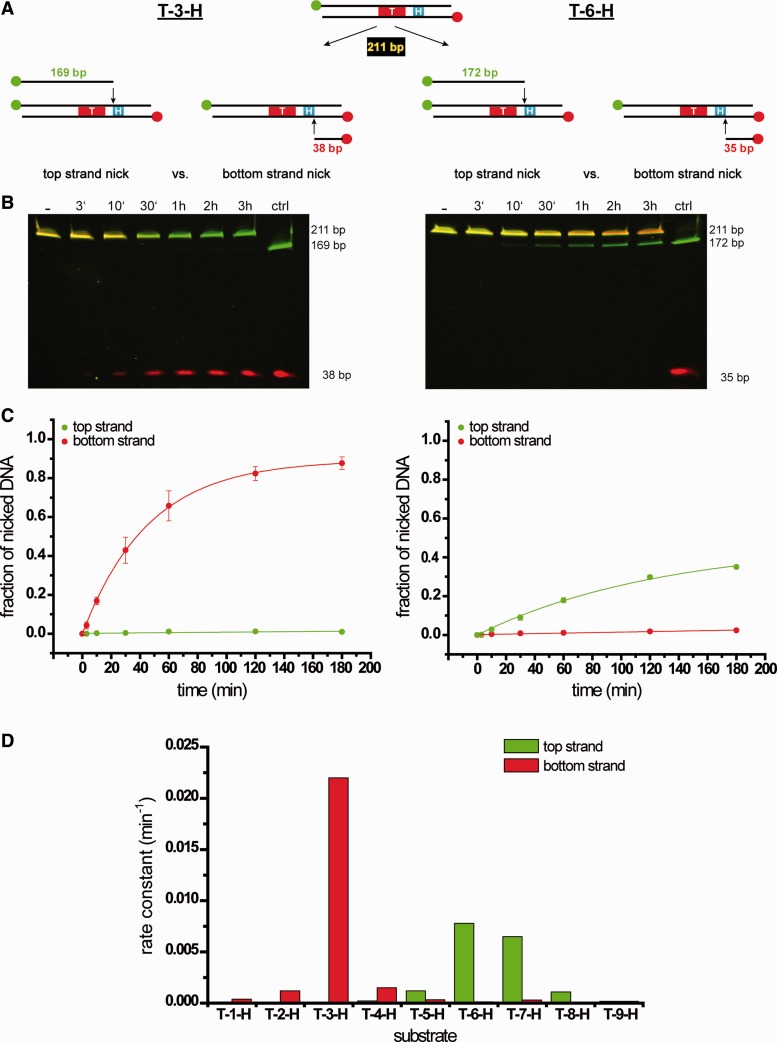
Targeted genome engineering requires nucleases that introduce a highly specific double-strand break in the genome that is either processed by homology-directed repair in the presence of a homologous repair template or by non-homologous end-joining (NHEJ) that usually results in insertions or deletions. The error-prone NHEJ can be efficiently suppressed by 'nickases' that produce a single-strand break rather than a double-strand break. Highly specific nickases have been produced by engineering of homing endonucleases and more recently by modifying zinc finger nucleases (ZFNs) composed of a zinc finger array and the catalytic domain of the restriction endonuclease FokI. These ZF-nickases work as heterodimers in which one subunit has a catalytically inactive FokI domain. We present two different approaches to engineer highly specific nickases; both rely on the sequence-specific nicking activity of the DNA mismatch repair endonuclease MutH which we fused to a DNA-binding module, either a catalytically inactive variant of the homing endonuclease I-SceI or the DNA-binding domain of the TALE protein AvrBs4. The fusion proteins nick strand specifically a bipartite recognition sequence consisting of the MutH and the I-SceI or TALE recognition sequences, respectively, with a more than 1000-fold preference over a stand-alone MutH site. TALE-MutH is a programmable nickase.
Figures
Similar articles
-
Creating highly specific nucleases by fusion of active restriction endonucleases and catalytically inactive homing endonucleases.Nucleic Acids Res. 2012 Jan;40(2):847-60. doi: 10.1093/nar/gkr788. Epub 2011 Sep 29. Nucleic Acids Res. 2012. PMID: 21965534 Free PMC article.
-
Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme.Genome Res. 2012 Jul;22(7):1316-26. doi: 10.1101/gr.122879.111. Epub 2012 Mar 20. Genome Res. 2012. PMID: 22434427 Free PMC article.
-
Chimeras of the homing endonuclease PI-SceI and the homologous Candida tropicalis intein: a study to explore the possibility of exchanging DNA-binding modules to obtain highly specific endonucleases with altered specificity.Chembiochem. 2004 Feb 6;5(2):206-13. doi: 10.1002/cbic.200300718. Chembiochem. 2004. PMID: 14760742
-
The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair.DNA Repair (Amst). 2016 Aug;44:6-16. doi: 10.1016/j.dnarep.2016.05.001. Epub 2016 May 12. DNA Repair (Amst). 2016. PMID: 27261202 Free PMC article. Review.
-
Homing endonucleases: from basics to therapeutic applications.Cell Mol Life Sci. 2010 Mar;67(5):727-48. doi: 10.1007/s00018-009-0188-y. Cell Mol Life Sci. 2010. PMID: 19915993 Free PMC article. Review.
Cited by
-
Visualizing DNA single- and double-strand breaks in the Flash comet assay by DNA polymerase-assisted end-labelling.Nucleic Acids Res. 2024 Feb 28;52(4):e22. doi: 10.1093/nar/gkae009. Nucleic Acids Res. 2024. PMID: 38261985 Free PMC article.
-
Performance of the Cas9 nickase system in Drosophila melanogaster.G3 (Bethesda). 2014 Aug 15;4(10):1955-62. doi: 10.1534/g3.114.013821. G3 (Bethesda). 2014. PMID: 25128437 Free PMC article.
-
Targeted mutagenesis: A sniper-like diversity generator in microbial engineering.Synth Syst Biotechnol. 2017 Jul 14;2(2):75-86. doi: 10.1016/j.synbio.2017.07.001. eCollection 2017 Jun. Synth Syst Biotechnol. 2017. PMID: 29062964 Free PMC article. Review.
-
Strand-selective base editing of human mitochondrial DNA using mitoBEs.Nat Biotechnol. 2024 Mar;42(3):498-509. doi: 10.1038/s41587-023-01791-y. Epub 2023 May 22. Nat Biotechnol. 2024. PMID: 37217751 Free PMC article.
-
Type II restriction endonucleases--a historical perspective and more.Nucleic Acids Res. 2014 Jul;42(12):7489-527. doi: 10.1093/nar/gku447. Epub 2014 May 30. Nucleic Acids Res. 2014. PMID: 24878924 Free PMC article. Review.
References
-
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010;11:636–646. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
