

Evolution and impact of subclonal mutations in chronic lymphocytic leukemia
- PMID: 23415222
- PMCID: PMC3575604
- DOI: 10.1016/j.cell.2013.01.019
Evolution and impact of subclonal mutations in chronic lymphocytic leukemia
Abstract
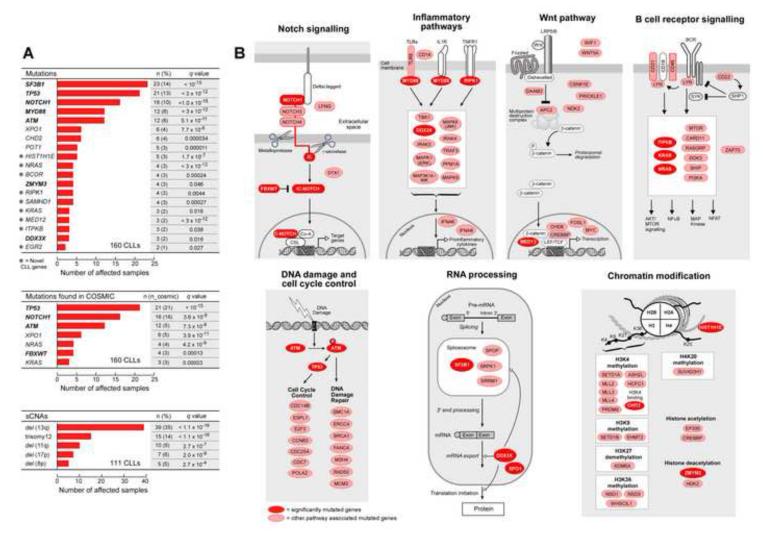
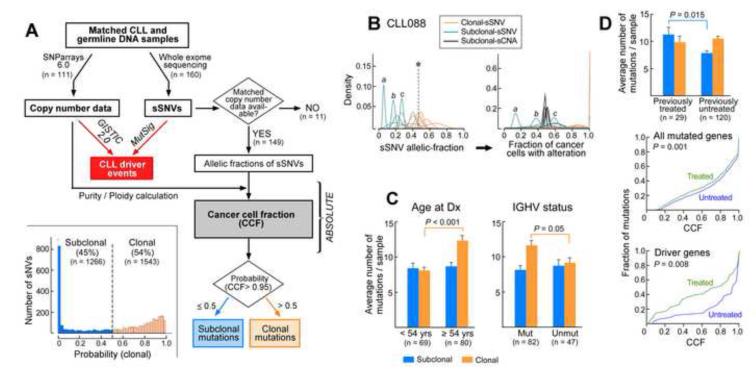
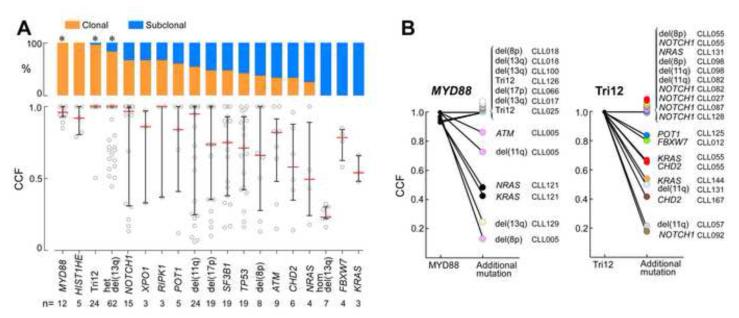
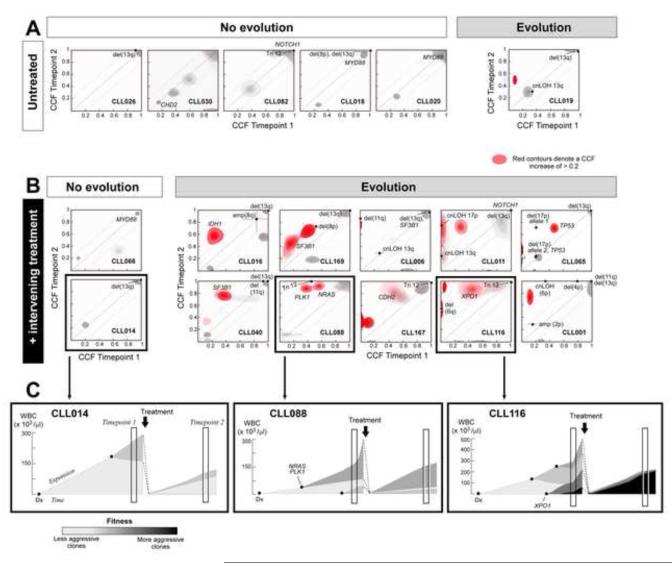
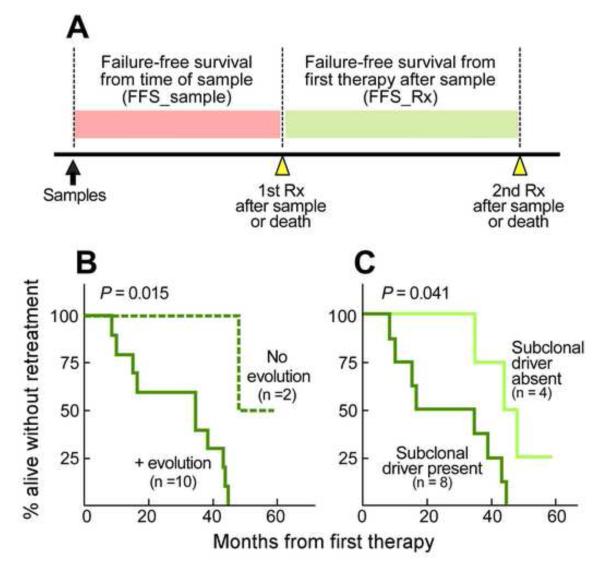
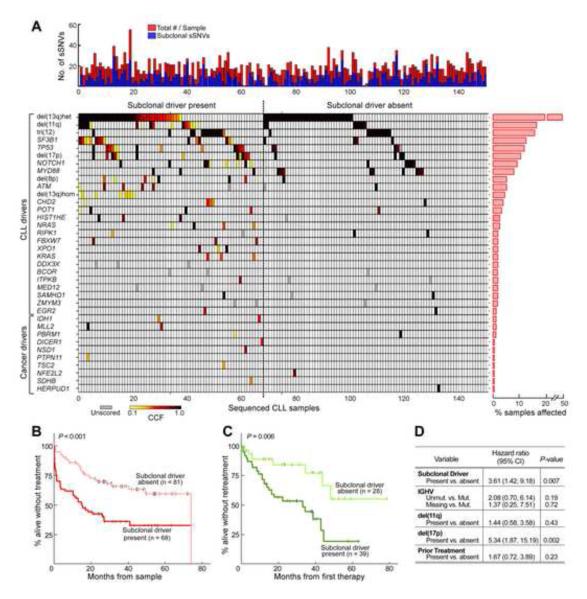
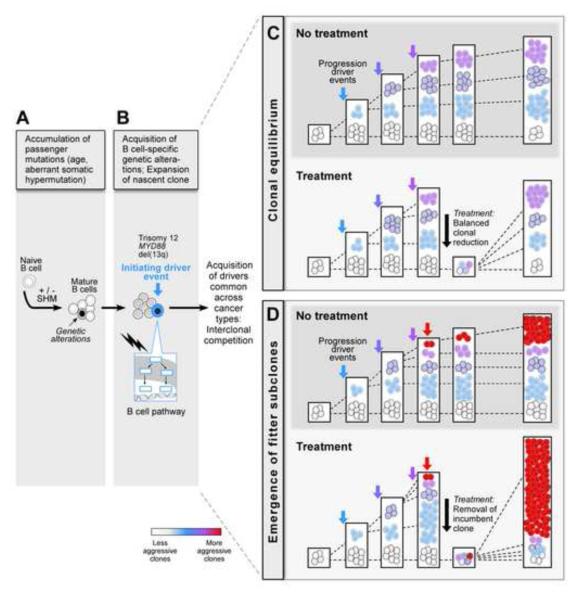
Clonal evolution is a key feature of cancer progression and relapse. We studied intratumoral heterogeneity in 149 chronic lymphocytic leukemia (CLL) cases by integrating whole-exome sequence and copy number to measure the fraction of cancer cells harboring each somatic mutation. We identified driver mutations as predominantly clonal (e.g., MYD88, trisomy 12, and del(13q)) or subclonal (e.g., SF3B1 and TP53), corresponding to earlier and later events in CLL evolution. We sampled leukemia cells from 18 patients at two time points. Ten of twelve CLL cases treated with chemotherapy (but only one of six without treatment) underwent clonal evolution, predominantly involving subclones with driver mutations (e.g., SF3B1 and TP53) that expanded over time. Furthermore, presence of a subclonal driver mutation was an independent risk factor for rapid disease progression. Our study thus uncovers patterns of clonal evolution in CLL, providing insights into its stepwise transformation, and links the presence of subclones with adverse clinical outcomes.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Tumour heterogeneity: The rise of the minority.Nat Rev Cancer. 2013 Apr;13(4):225. doi: 10.1038/nrc3499. Nat Rev Cancer. 2013. PMID: 23518702 No abstract available.
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995;57:289–300.
-
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
