

Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging
- PMID: 23415915
- PMCID: PMC12163706
- DOI: 10.1016/j.stem.2013.01.017
Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging
Abstract
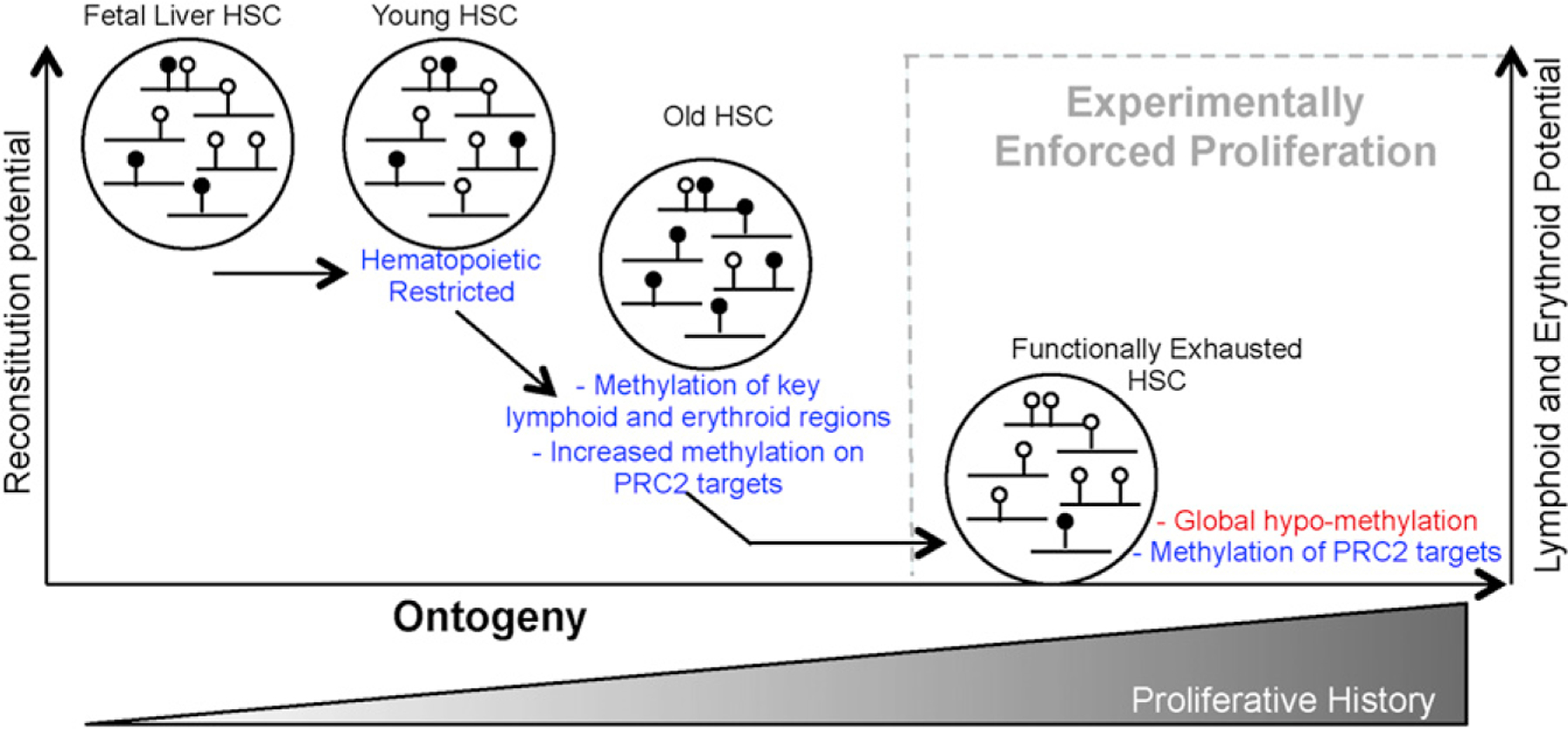
The functional potential of hematopoietic stem cells (HSCs) declines during aging, and in doing so, significantly contributes to hematopoietic pathophysiology in the elderly. To explore the relationship between age-associated HSC decline and the epigenome, we examined global DNA methylation of HSCs during ontogeny in combination with functional analysis. Although the DNA methylome is generally stable during aging, site-specific alterations of DNA methylation occur at genomic regions associated with hematopoietic lineage potential and selectively target genes expressed in downstream progenitor and effector cells. We found that age-associated HSC decline, replicative limits, and DNA methylation are largely dependent on the proliferative history of HSCs, yet appear to be telomere-length independent. Physiological aging and experimentally enforced proliferation of HSCs both led to DNA hypermethylation of genes regulated by Polycomb Repressive Complex 2. Our results provide evidence that epigenomic alterations of the DNA methylation landscape contribute to the functional decline of HSCs during aging.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures






References
-
- Adolfsson J, Månsson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, et al. (2005). Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 121, 295–306. - PubMed
-
- Allsopp RC, Morin GB, Horner JW, DePinho R, Harley CB, and Weissman IL (2003). Effect of TERT over-expression on the long-term transplantation capacity of hematopoietic stem cells. Nat. Med 9, 369–371. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
