

Flexible backbone sampling methods to model and design protein alternative conformations
- PMID: 23422426
- PMCID: PMC3750959
- DOI: 10.1016/B978-0-12-394292-0.00004-7
Flexible backbone sampling methods to model and design protein alternative conformations
Abstract
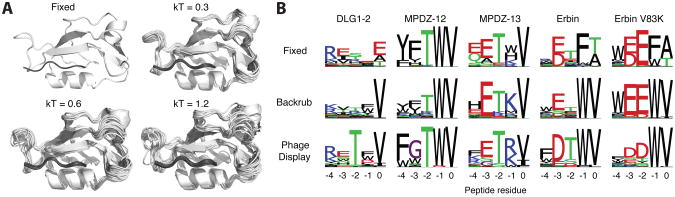
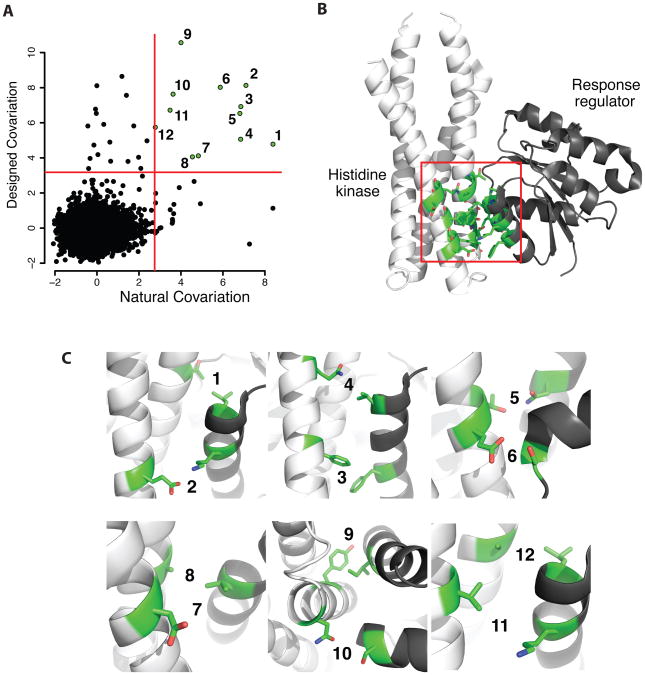
Sampling alternative conformations is key to understanding how proteins work and engineering them for new functions. However, accurately characterizing and modeling protein conformational ensembles remain experimentally and computationally challenging. These challenges must be met before protein conformational heterogeneity can be exploited in protein engineering and design. Here, as a stepping stone, we describe methods to detect alternative conformations in proteins and strategies to model these near-native conformational changes based on backrub-type Monte Carlo moves in Rosetta. We illustrate how Rosetta simulations that apply backrub moves improve modeling of point mutant side-chain conformations, native side-chain conformational heterogeneity, functional conformational changes, tolerated sequence space, protein interaction specificity, and amino acid covariation across protein-protein interfaces. We include relevant Rosetta command lines and RosettaScripts to encourage the application of these types of simulations to other systems. Our work highlights that critical scoring and sampling improvements will be necessary to approximate conformational landscapes. Challenges for the future development of these methods include modeling conformational changes that propagate away from designed mutation sites and modulating backbone flexibility to predictively design functionally important conformational heterogeneity.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
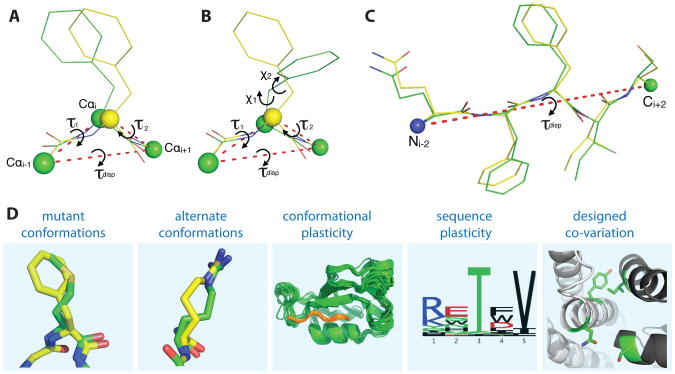
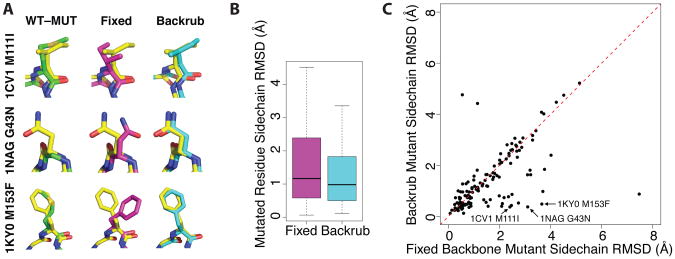
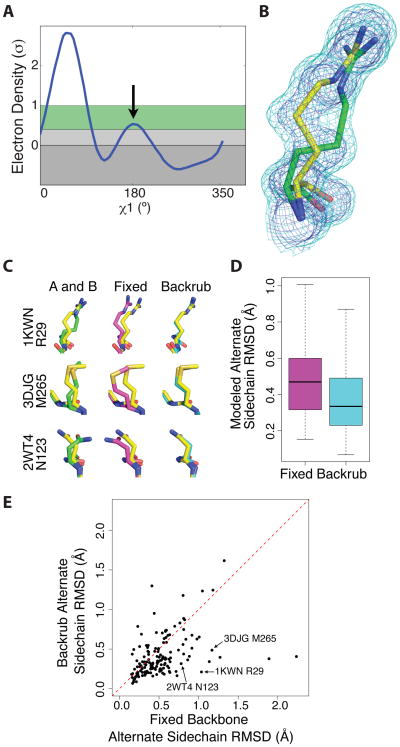
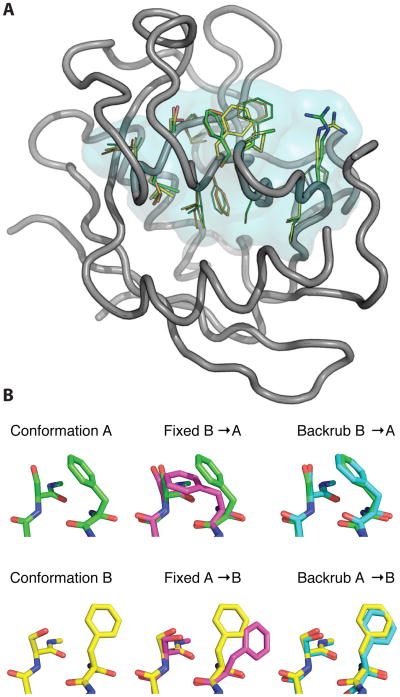
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
