

Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study
- PMID: 23424287
- PMCID: PMC3570532
- DOI: 10.1371/journal.pmed.1001387
Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study
Abstract
Background: Understanding Mycobacterium tuberculosis (Mtb) transmission is essential to guide efficient tuberculosis control strategies. Traditional strain typing lacks sufficient discriminatory power to resolve large outbreaks. Here, we tested the potential of using next generation genome sequencing for identification of outbreak-related transmission chains.
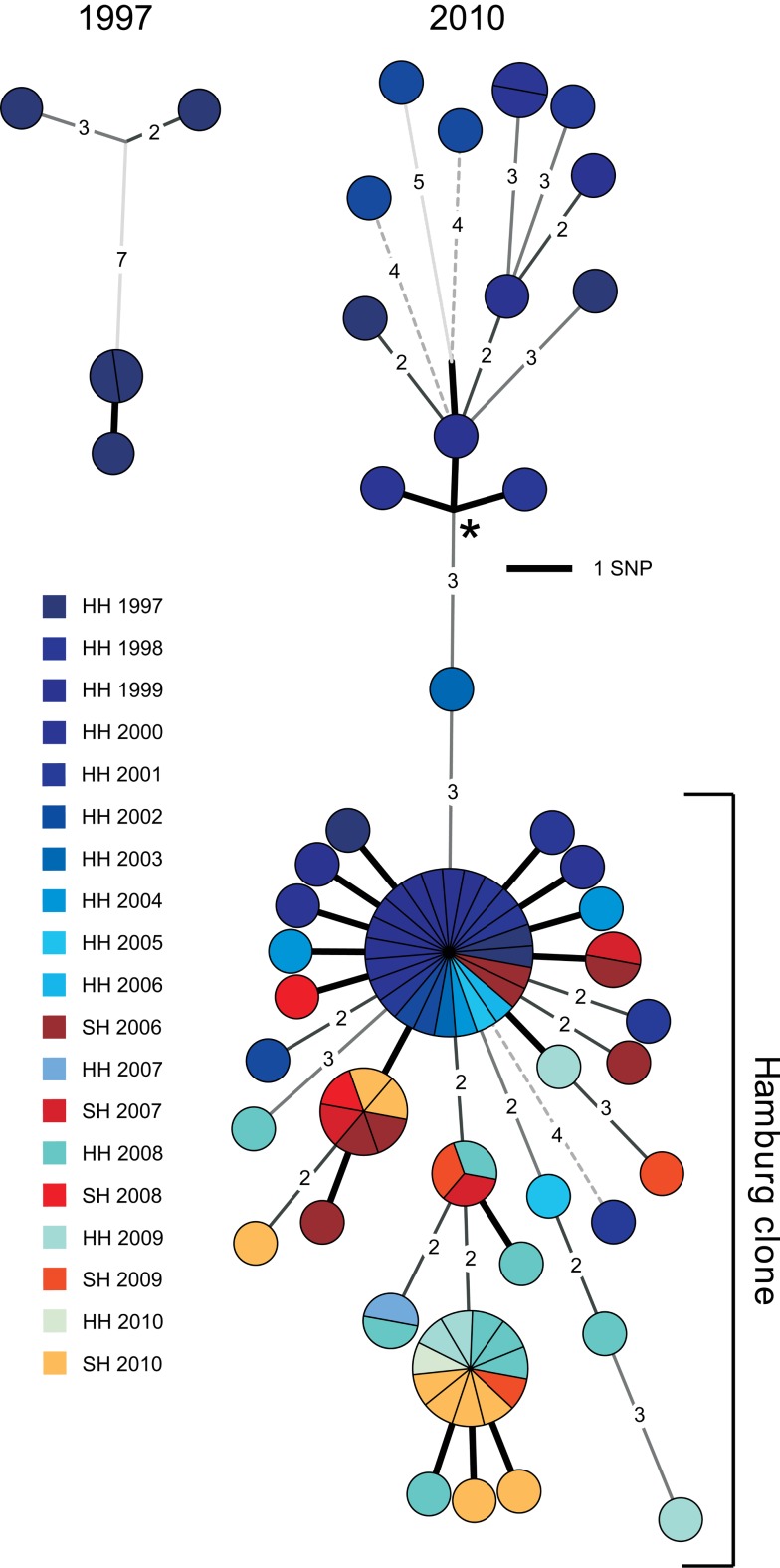
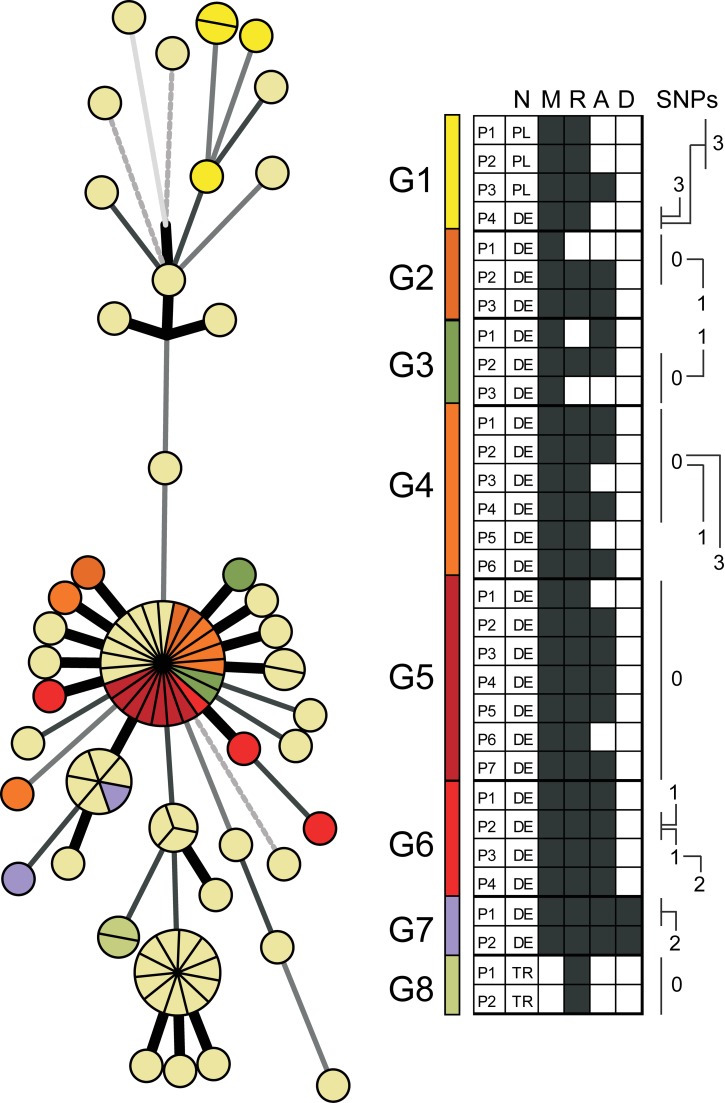
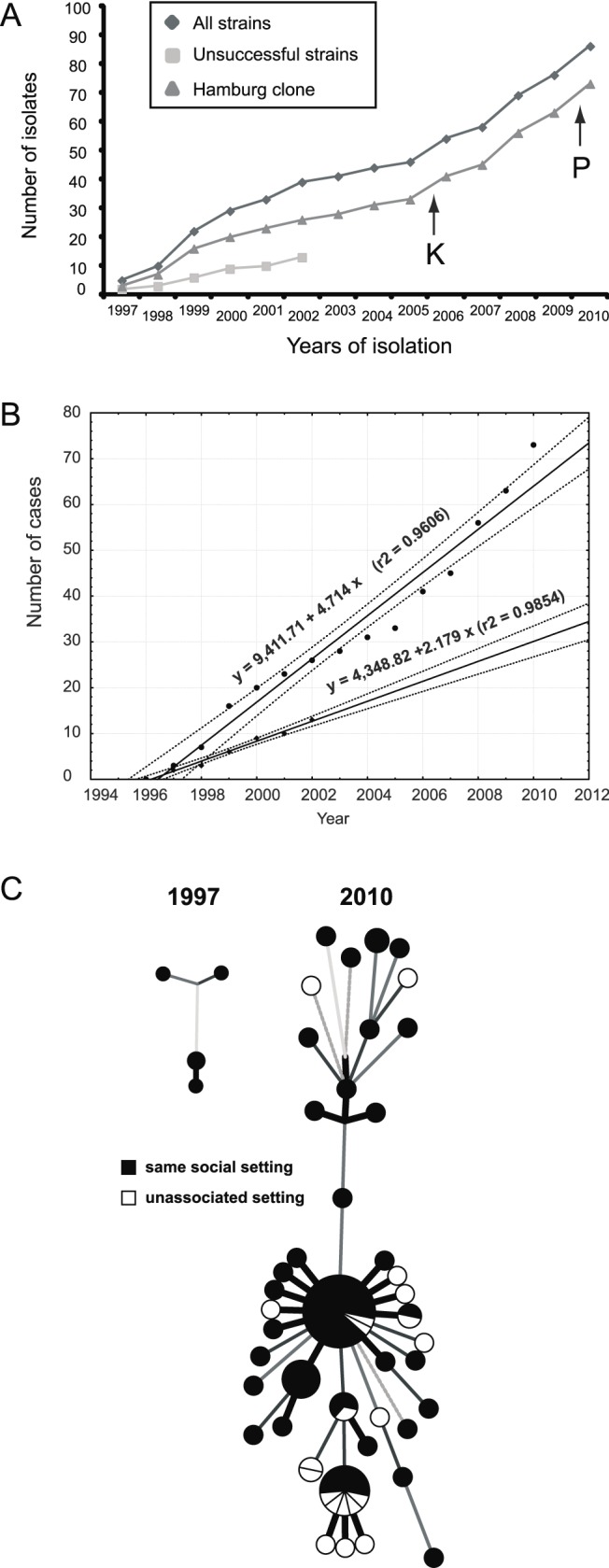
Methods and findings: During long-term (1997 to 2010) prospective population-based molecular epidemiological surveillance comprising a total of 2,301 patients, we identified a large outbreak caused by an Mtb strain of the Haarlem lineage. The main performance outcome measure of whole genome sequencing (WGS) analyses was the degree of correlation of the WGS analyses with contact tracing data and the spatio-temporal distribution of the outbreak cases. WGS analyses of the 86 isolates revealed 85 single nucleotide polymorphisms (SNPs), subdividing the outbreak into seven genome clusters (two to 24 isolates each), plus 36 unique SNP profiles. WGS results showed that the first outbreak isolates detected in 1997 were falsely clustered by classical genotyping. In 1998, one clone (termed "Hamburg clone") started expanding, apparently independently from differences in the social environment of early cases. Genome-based clustering patterns were in better accordance with contact tracing data and the geographical distribution of the cases than clustering patterns based on classical genotyping. A maximum of three SNPs were identified in eight confirmed human-to-human transmission chains, involving 31 patients. We estimated the Mtb genome evolutionary rate at 0.4 mutations per genome per year. This rate suggests that Mtb grows in its natural host with a doubling time of approximately 22 h (400 generations per year). Based on the genome variation discovered, emergence of the Hamburg clone was dated back to a period between 1993 and 1997, hence shortly before the discovery of the outbreak through epidemiological surveillance.
Conclusions: Our findings suggest that WGS is superior to conventional genotyping for Mtb pathogen tracing and investigating micro-epidemics. WGS provides a measure of Mtb genome evolution over time in its natural host context.
Conflict of interest statement
PS is a consultant for Genoscreen. All other authors have declared that no competing interests exist.
Figures
References
-
- Dye C, Williams BG (2010) The population dynamics and control of tuberculosis. Science 328: 856–861 doi:10.1126/science.1185449. - DOI - PubMed
-
- World Health Organization (2012) Global tuberculosis report 2012. Available: http://www.who.int/tb/publications/global_report/en/index.html. Accessed 8 January 2013.
-
- Read AF (1994) The evolution of virulence. Trends Microbiol 2: 73–76. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
