

Genetics of the epilepsies: where are we and where are we going?
- PMID: 23429546
- PMCID: PMC3781236
- DOI: 10.1097/WCO.0b013e32835ee6ff
Genetics of the epilepsies: where are we and where are we going?
Abstract
Purpose of review: We aim to review the most recent advances in the field of epilepsy genetics with particular focus on the progress in gene discovery in monogenic epilepsies, identification of risk genes in complex genetic epilepsies and recent findings in the field of epilepsy pharmacogenomics.
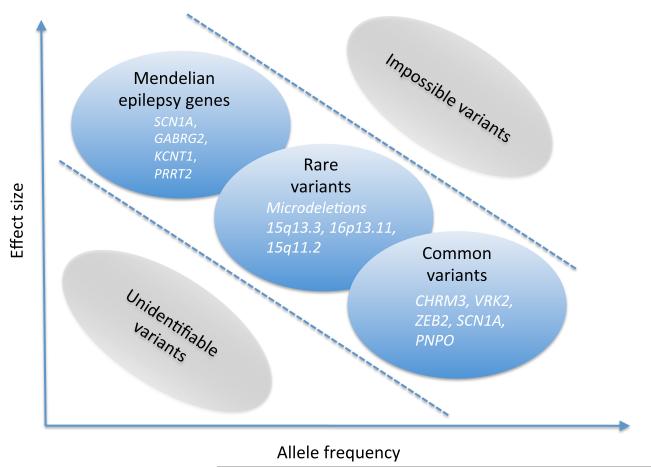
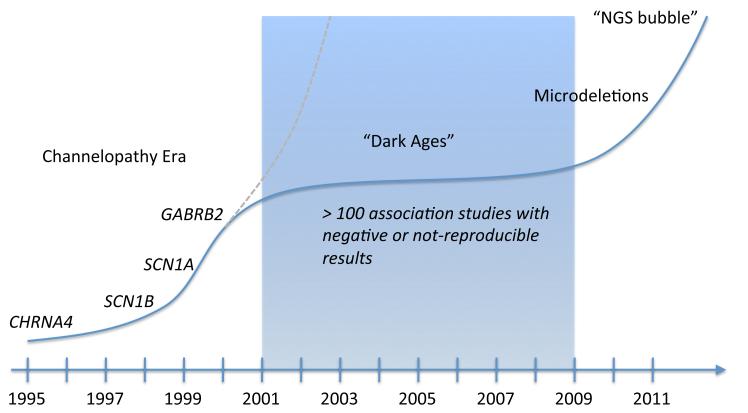
Recent findings: During the last 12 months, the use of massive parallel sequencing technologies has allowed for the discovery of several genes for monogenic epilepsies. Most importantly, PRRT2 was identified as the long-sought gene for benign familial infantile seizures. Mutations in KCNT1 were found in two seemingly unrelated monogenic epilepsies including malignant migrating partial seizures of infancy and severe autosomal dominant nocturnal frontal lobe epilepsy. A genome-wide association study in idiopathic generalized epilepsy revealed the first common risk variants for human seizure disorders including variants in VRK2, PNPO and SCN1A. Furthermore, a landmark study provided evidence that screening for the HLA-B1502 variant may prevent carbamazepine CBZ-induced side effects in the Taiwanese population. Also, HLA-A3101 variants were identified as a risk factor for carbamazepine side effects in Europeans.
Summary: Novel technologies and an unprecedented level of international collaboration have resulted in identification of novel genes for monogenic and complex genetic epilepsies as well as risk factors for side effects of antiepileptic drugs. This review provides an overview of the most relevant studies in the last year and highlights the future direction of the field.
Figures
References
-
-
Epi4K: gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–67. * This review provides an overview of the Epi4K consortium and outlines the workflow of gene discovery in the era of high-throughput genomics.
-
-
- Claes L, Ceulemans B, Audenaert D, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. 2003;21:615–21. - PubMed
-
- Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843–52. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
