

Subtype-specific estrogen receptor-mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat
- PMID: 23429596
- PMCID: PMC3664271
- DOI: 10.1097/FJC.0b013e31828bc88a
Subtype-specific estrogen receptor-mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat
Abstract
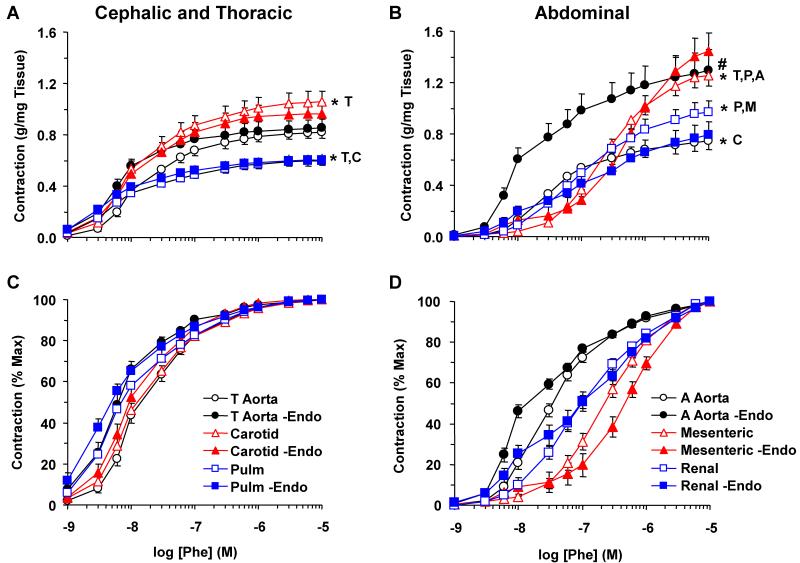
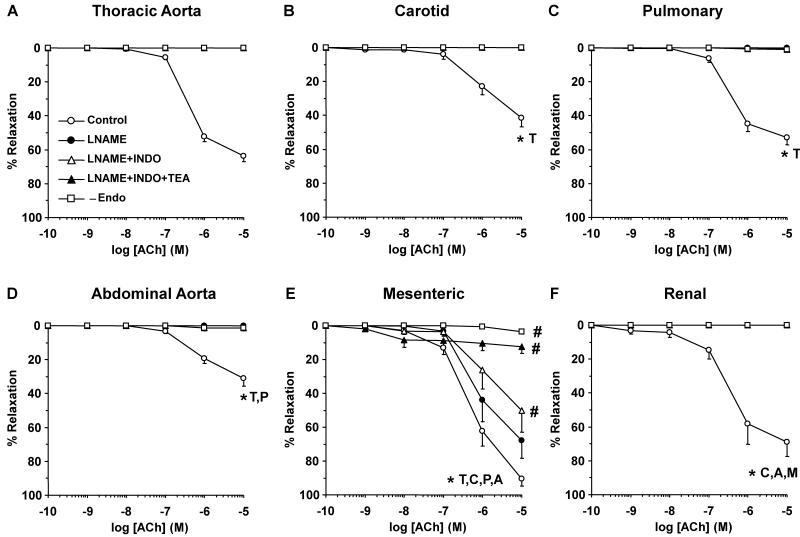
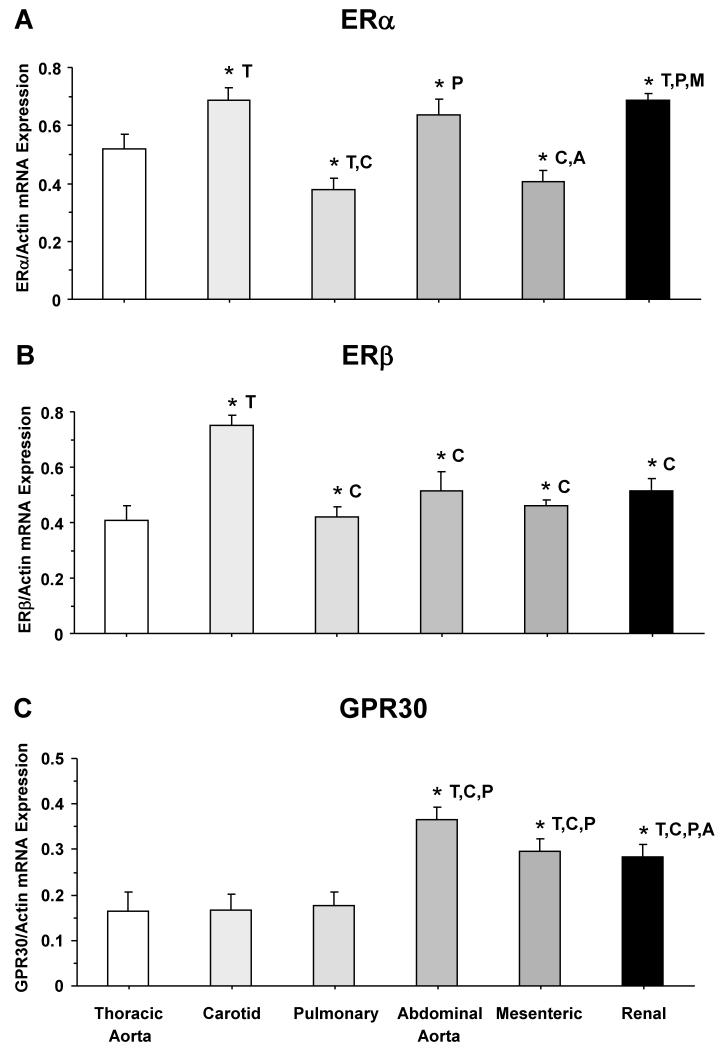
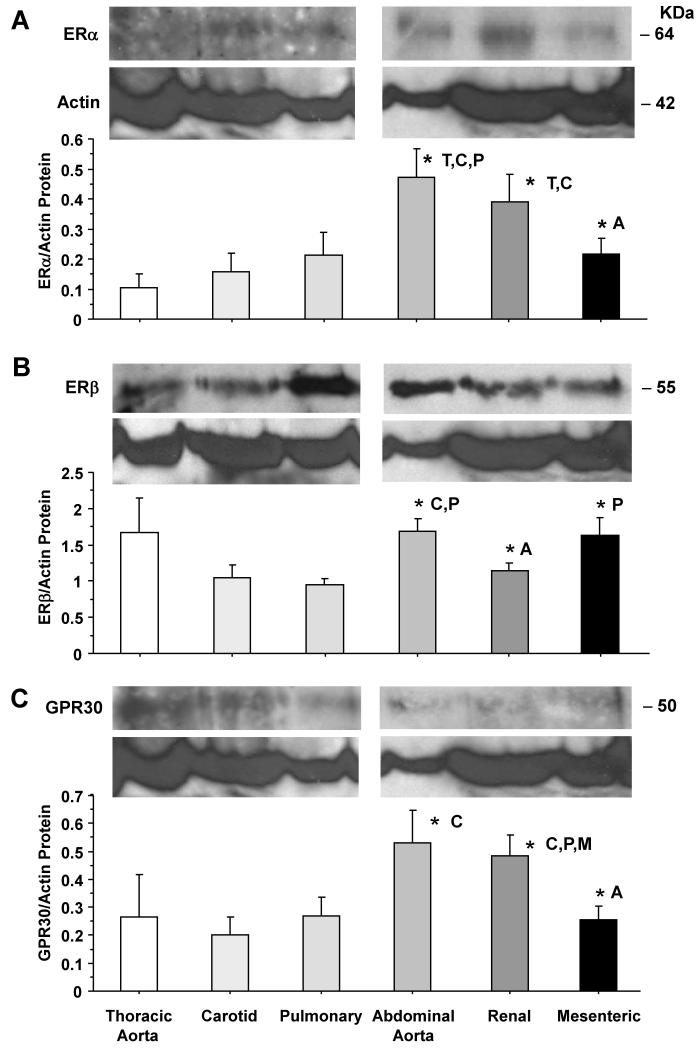
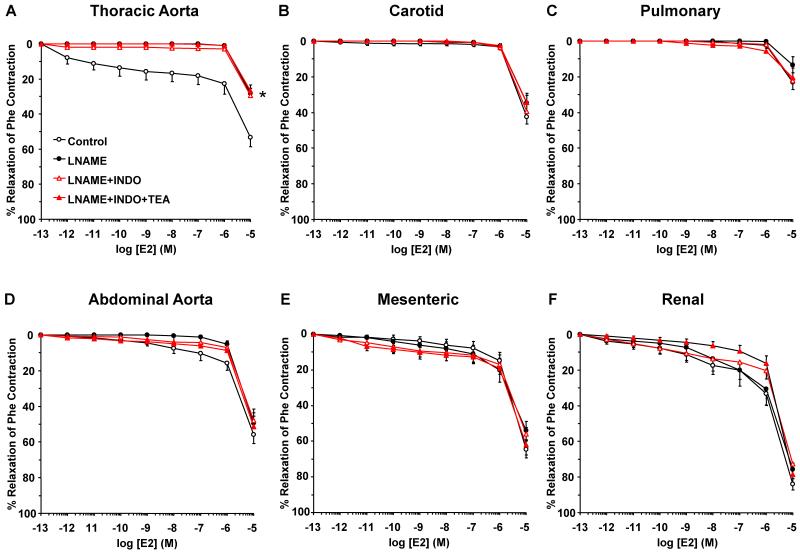
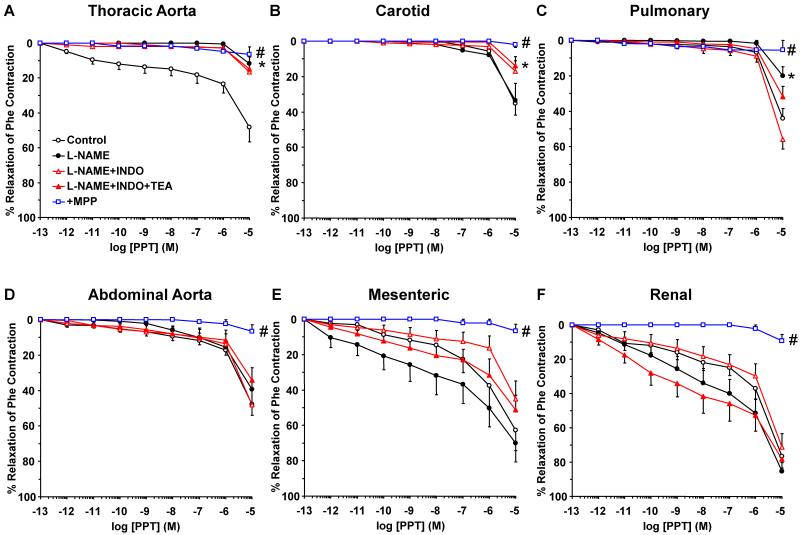
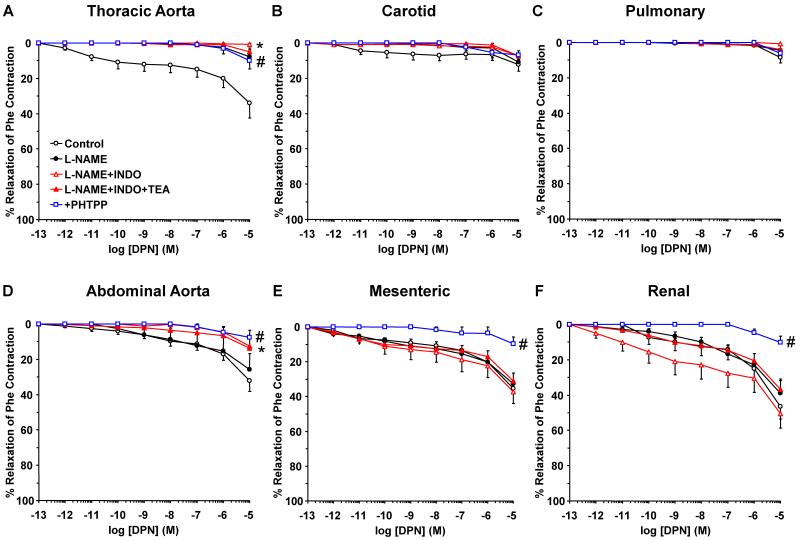
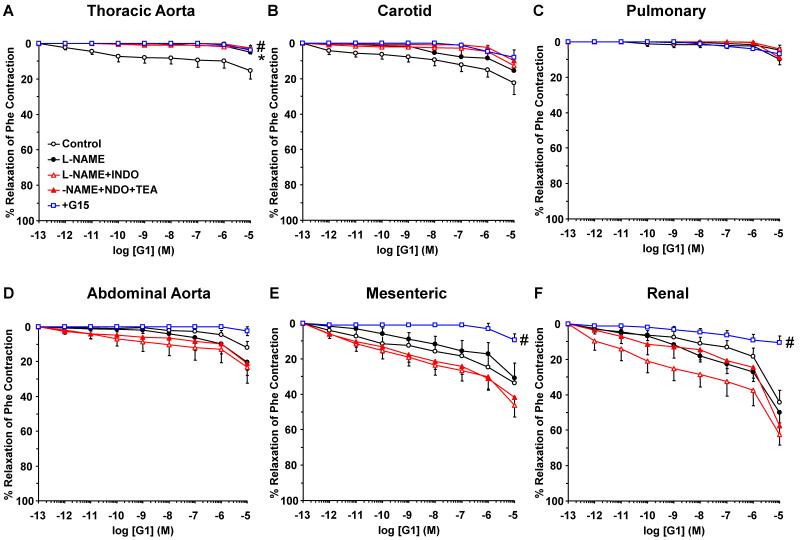
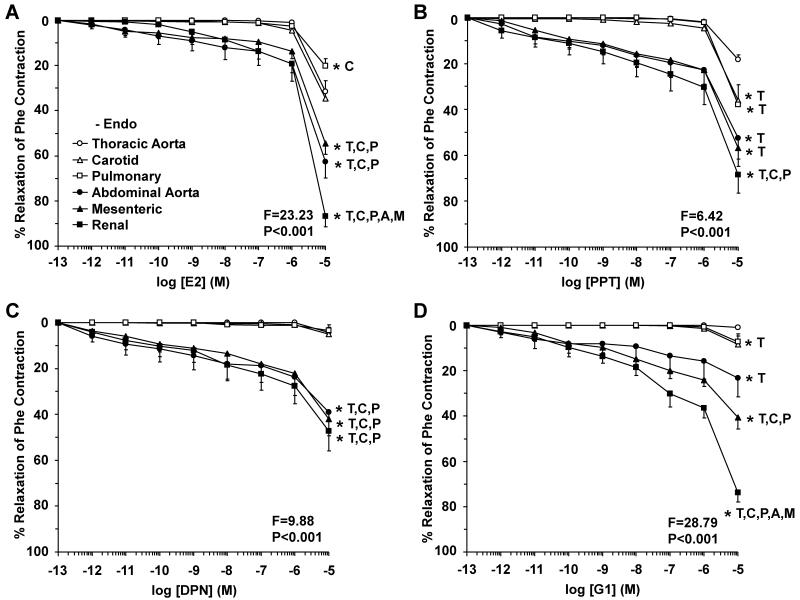
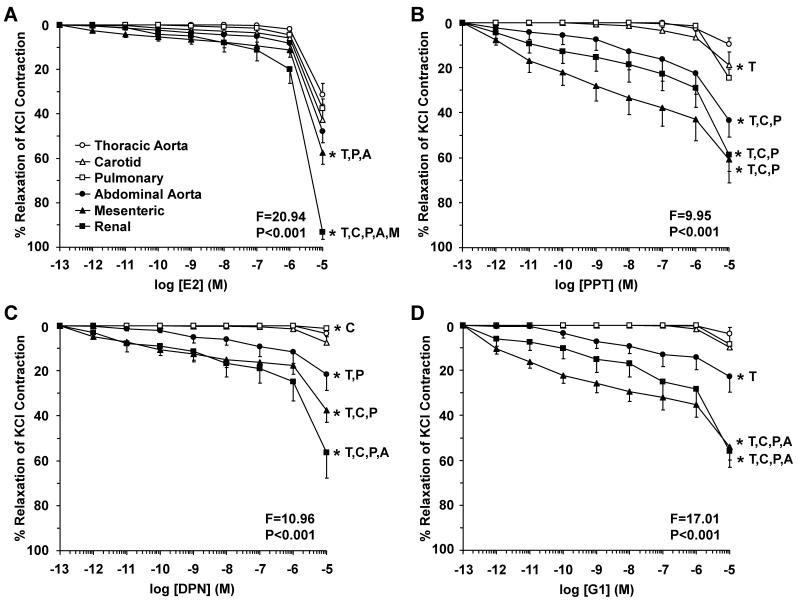
Estrogen receptors (ERs) mediate genomic and nongenomic vasodilator effects, but estrogen therapy may not provide systemic vascular protection. To test whether this is because of regional differences in ER distribution or vasodilator activity, cephalic (carotid artery), thoracic (thoracic aorta and pulmonary artery), and abdominal arteries (abdominal aorta, mesenteric artery, and renal artery) from female Sprague-Dawley rats were prepared to measure contraction to phenylephrine and relaxation to acetylcholine (ACh) and the ER activators 17β-estradiol (E2) (all ERs), 4,4',4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl)-tris-phenol (PPT) (ERα), diarylpropionitrile (DPN) (ERβ), and (±)-1-[(3aR*,4S*,9bS*)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone (G1) (GPR30). Phenylephrine caused contraction that was enhanced in endothelium-denuded aorta, supporting endothelial release of vasodilators. In cephalic and thoracic arteries, ACh relaxation was abolished by the nitric oxide (NO) synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME), suggesting a role of NO. In mesenteric vessels, ACh-induced relaxation was partly inhibited by the L-NAME + cyclooxygenase inhibitor indomethacin and blocked by the K+ channel blocker tetraethylammonium, suggesting a hyperpolarization pathway. E2 and PPT caused similar relaxation in all vessels. DPN and G1 caused smaller relaxation that was more prominent in abdominal vessels. Reverse transcription-polymerase chain reaction revealed variable ERα messenger RNA expression and increased ERβ in carotid artery and GPR30 in abdominal arteries. Western blots revealed greater amounts of ERα, ERβ, and GPR30 in abdominal arteries. In thoracic aorta, E2-, PPT-, and DPN-induced relaxation was blocked by L-NAME and was associated with increased nitrite/nitrate production, suggesting a role of NO. In abdominal vessels, E2-, PPT-, DPN-, and G1-induced relaxation persisted in L-NAME + indomethacin + tetraethylammonium-treated or endothelium-denuded arteries, suggesting direct effect on vascular smooth muscle. E2, PPT, DPN, and G1 caused greater relaxation of KCl-induced contraction in abdominal vessels, suggesting inhibitory effects on Ca2+ entry. Thus, E2 and ERα stimulation produces similar relaxation of the cephalic, thoracic, and abdominal arteries. In the cephalic and thoracic arteries, particularly the thoracic aorta, E2-induced and ERα- and ERβ-mediated vasodilation involves NO production. ERβ- and GPR30-mediated relaxation is greater in the abdominal arteries and seems to involve hyperpolarization and inhibition of vascular smooth muscle Ca2+ entry. Specific ER agonists could produce vasodilation in specific vascular beds without affecting other vessels in the systemic circulation.
Figures


References
-
- Barrett-Connor E, Bush TL. Estrogen and coronary heart disease in women. JAMA. 1991;265(14):1861–1867. - PubMed
-
- Gerhard M, Ganz P. How do we explain the clinical benefits of estrogen? From bedside to bench. Circulation. 1995;92(1):5–8. - PubMed
-
- Dubey RK, Imthurn B, Zacharia LC, Jackson EK. Hormone replacement therapy and cardiovascular disease: what went wrong and where do we go from here? Hypertension. 2004;44(6):789–795. - PubMed
-
- Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol. 2004;286(2):R233–249. - PubMed
-
- Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308(5728):1583–1587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
