

5-Oxoprolinuria in Heterozygous Patients for 5-Oxoprolinase (OPLAH) Missense Changes
- PMID: 23430506
- PMCID: PMC3575052
- DOI: 10.1007/8904_2012_166
5-Oxoprolinuria in Heterozygous Patients for 5-Oxoprolinase (OPLAH) Missense Changes
Abstract
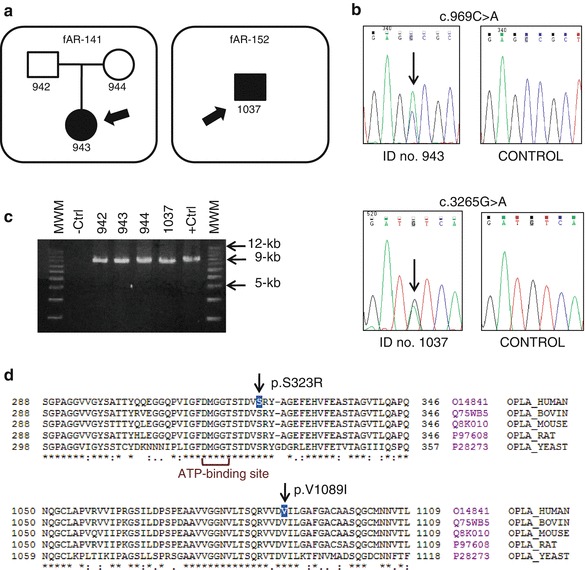
The inherited 5-oxoprolinuria is primarily suggestive of genetic defects in two enzymes belonging to the gamma-glutamyl cycle in the glutathione (GSH) metabolism: the glutathione synthetase (GSS) and the 5-oxoprolinase (OPLAH). The GSS deficiency is the best characterized of the inborn errors of GSH metabolism, whereas the OPLAH deficiency is questioned whether it is a disorder or just a biochemical condition with no adverse clinical effects. Recently, the first human OPLAH mutation (p.H870Pfs) was reported in homozygosis in two siblings who suffered from 5-oxoprolinuria with a benign clinical course. We report two unrelated patients who manifested massive excretion of 5-oxoproline in urine. In both probands, the blood GSH levels were normal and no mutations were found in the GSS gene. The mutational screening of the OPLAH gene, which included the codified sequences, the intronic flanking sequences, the promoter sequence, and a genetic analysis in order to detect large deletions and/or duplications, showed that each patient only harbors one missense mutation in heterozygosis. The in silico analyses revealed that each one of these OPLAH mutations, p.S323R and p.V1089I, could alter the proper function of this homodimeric enzyme. In addition, clinical symptoms manifest in these two probands were not related to GSH cycle defects and, therefore, this study provides further evidence that oxoprolinuria may present as epiphenomenon in several pathological conditions and confound the final diagnosis.
Figures
References
-
- Almaghlouth I, Mohamed J, Al-Amoudi M, Al-Ahaidib L, Al-Odaib A, Alkuraya F (2011) 5-Oxoprolinase deficiency: report of the first human OPLAH mutation. Clin Genet. doi:10.1111/j.1399-0004.2011.01728.x - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Benson MD. The metabolic and molecular bases of inherited disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Amyloidosis. New York: McGraw-Hill; 2001. pp. 5345–5378.
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
