

Further Delineation of the Phenotype of Congenital Disorder of Glycosylation DPAGT1-CDG (CDG-Ij) Identified by Homozygosity Mapping
- PMID: 23430862
- PMCID: PMC3509848
- DOI: 10.1007/8904_2011_57
Further Delineation of the Phenotype of Congenital Disorder of Glycosylation DPAGT1-CDG (CDG-Ij) Identified by Homozygosity Mapping
Abstract
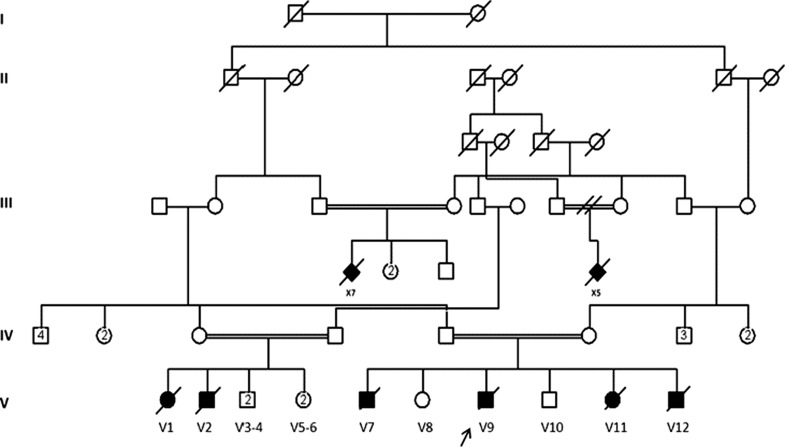
Congenital disorders of glycosylation (CDG) are an expanding group of genetic diseases affecting protein and lipid glycosylation. These disorders have a variable presentation and are individually rare. DPAGT1-CDG is a protein N-glycosylation disorder with epilepsy, development delay, severe hypotonia, and dysmorphy, reported in a single patient. Here we present the second family with DPAGT1-CDG identified through homozygosity mapping in a large consanguineous family with 18 affected infants. The patients had severe hypotonia, global developmental delay, seizures, and microcephaly but no dysmorphy. In the index case, the brain MRI revealed delayed myelination, and there was fiber type disproportion on muscle biopsy. Homozygosity mapping identified a large block of homozygosity on chromosome 11p15.5-q25 where two known CDG-I causing genes, ALG9 and DPAGT1, are located. Sequencing ALG9 did not reveal any mutations while analysis of DPAGT1 identified a novel homozygous mutation c.902G>A (p.R301H) in two affected infants. The disorder was fatal in all affected cases and mostly in early infancy.
Figures

References
-
- Clarke NF, North KN. Congenital fiber type disproportion–30 years on. J Neuropathol Exp Neurol. 2003;62:977–989. - PubMed
LinkOut - more resources
Full Text Sources
