

Insulin resistance and muscle metabolism in chronic kidney disease
- PMID: 23431467
- PMCID: PMC3575670
- DOI: 10.1155/2013/329606
Insulin resistance and muscle metabolism in chronic kidney disease
Abstract
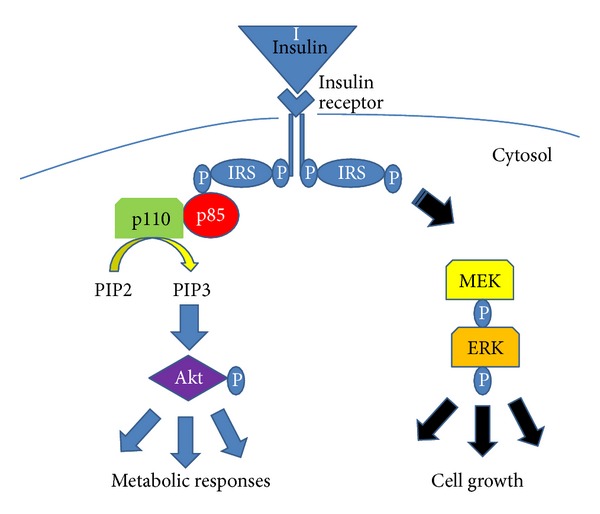
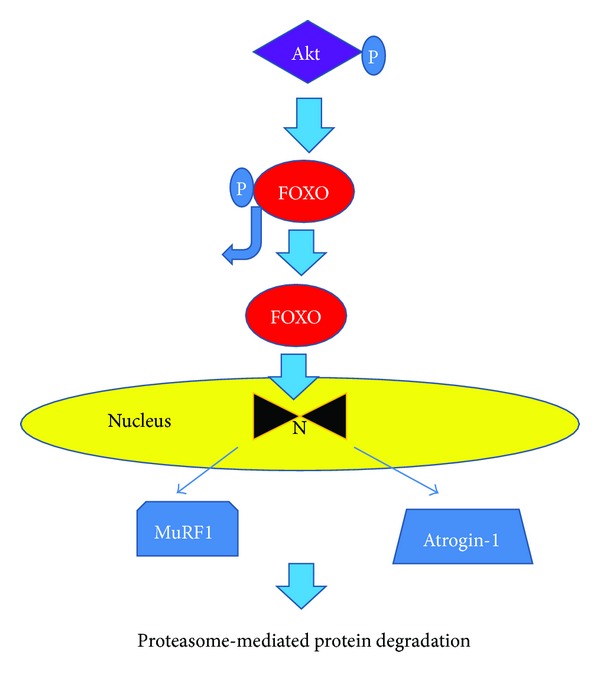
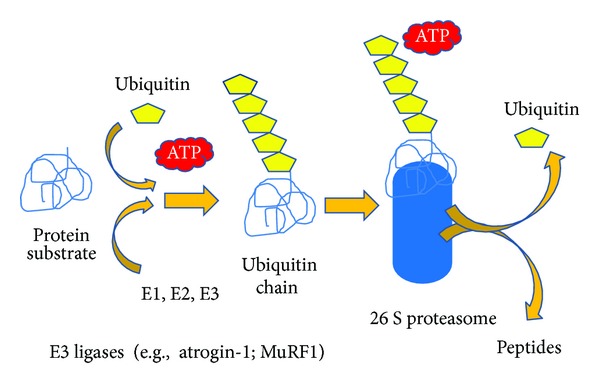
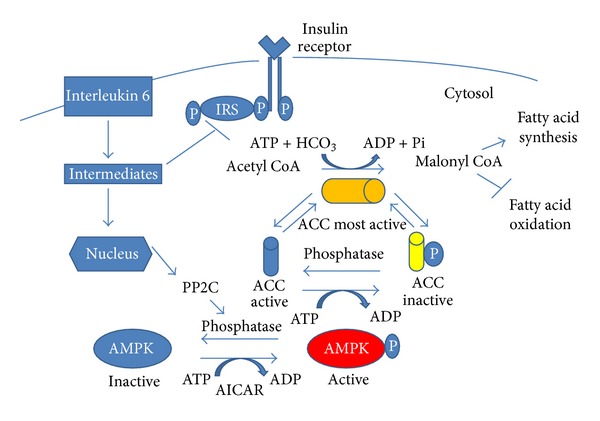
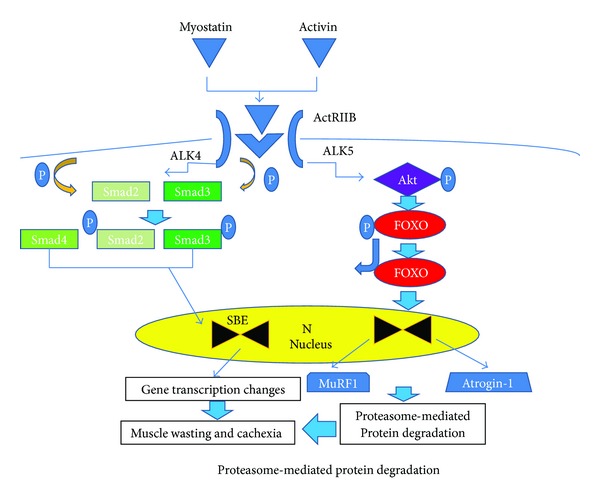
Insulin resistance is a common finding in chronic kidney disease (CKD) and is manifested by mild fasting hyperglycemia and abnormal glucose tolerance testing. Circulating levels of glucocorticoids are high. In muscle, changes in the insulin signaling pathway occur. An increase in the regulatory p85 subunit of Class I phosphatidylinositol 3-Kinase enzyme leads to decreased activation of the downstream effector protein kinase B (Akt). Mechanisms promoting muscle proteolysis and atrophy are unleashed. The link of Akt to the ubiquitin proteasome pathway, a major degradation pathway in muscle, is discussed. Another factor associated with insulin resistance in CKD is angiotensin II (Ang II) which appears to induce its intracellular effects through inflammatory cytokines or reactive oxygen species. Skeletal muscle ATP is depleted and the ability of AMP-activated protein kinase (AMPK) to replenish energy stores is blocked. How this can be reversed is discussed. Interleukin-6 (IL-6) levels are elevated in CKD and impair insulin signaling at the level of IRS-1. With exercise, IL-6 levels are reduced; glucose uptake and utilization are increased. For patients with CKD, exercise may improve insulin signaling and build up muscle. Treatment strategies for preventing muscle atrophy are discussed.
Figures
References
-
- Mak RHK, Haycock GB, Chantler C. Glucose intolerance in children with chronic renal failure. Kidney International. 1983;24(supplement 15):S22–S26. - PubMed
-
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effect of renal disease on renal uptake and excretion of insulin in man. New England Journal of Medicine. 1970;282(4):182–187. - PubMed
-
- Mondon CE, Dolkas CB, Reaven GM. Effect of acute uremia on insulin removal by the isolated perfused rat liver and muscle. Metabolism: Clinical and Experimental. 1978;27(2):133–142. - PubMed
-
- Cecchin F, Ittoop O, Sinha MK, Caro JF. Insulin resistance in uremia: insulin receptor kinase activity in liver and muscle from chronic uremic rats. American Journal of Physiology. 1988;254(4, part 1):E394–E401. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
