

DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition
- PMID: 23432899
- PMCID: PMC3605128
- DOI: 10.1186/1742-4690-10-21
DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition
Abstract
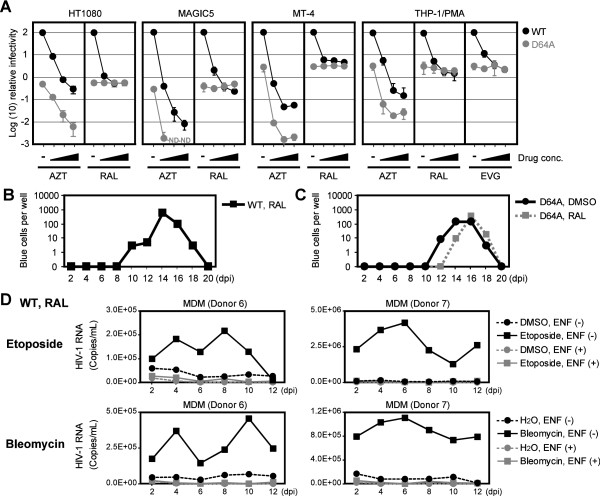
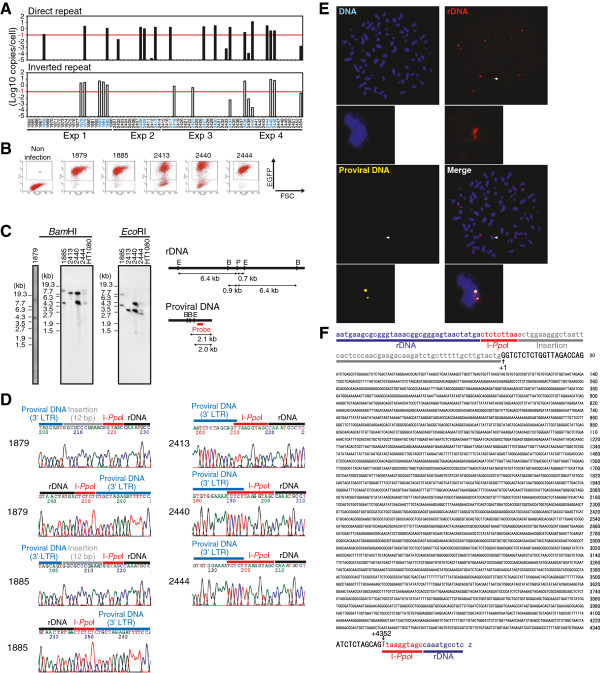
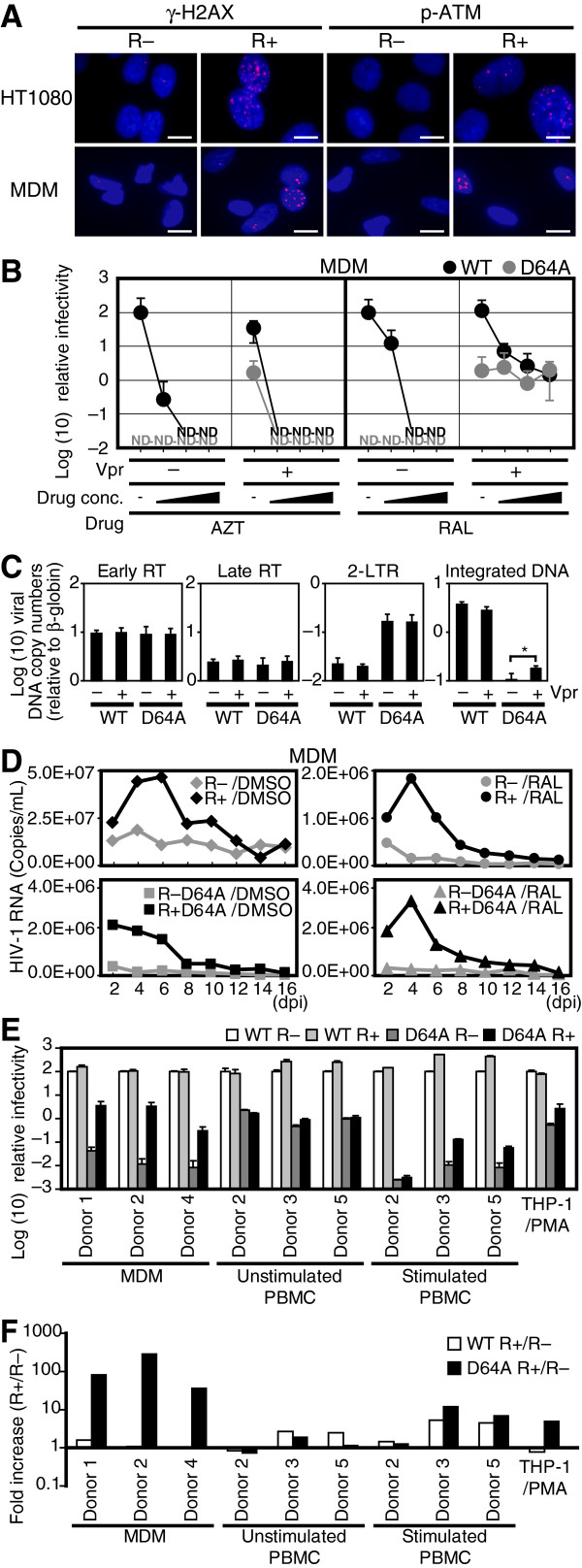
Background: The prevention of persistent human immunodeficiency virus type 1 (HIV-1) infection requires the clarification of the mode of viral transduction into resting macrophages. Recently, DNA double-strand breaks (DSBs) were shown to enhance infection by D64A virus, which has a defective integrase catalytic activity (IN-CA). However, the mechanism by which DSBs upregulate viral transduction was unclear. Here we analyzed the roles of DSBs during IN-CA-independent viral transduction into macrophages.
Results: We used cellular systems with rare-cutting endonucleases and found that D64A virus integrated efficiently into the sites of artificially induced DSBs. This IN-CA-independent viral transduction was blocked by an inhibitor of ataxia telangiectasia mutated protein (ATM) but was resistant to raltegravir (RAL), an inhibitor of integrase activity during strand transfer. Moreover, Vpr, an accessory gene product of HIV-1, induced DSBs in resting macrophages and significantly enhanced the rate of IN-CA-independent viral transduction into macrophages with concomitant production of secondary viruses.
Conclusion: DSBs contribute to the IN-CA-independent viral infection of macrophages, which is resistant to RAL. Thus, the ATM-dependent cellular pathway and Vpr-induced DNA damage are novel targets for preventing persistent HIV-1 infection.
Figures
Similar articles
-
HIV-1 Vpr-induced DNA damage activates NF-κB through ATM-NEMO independent of cell cycle arrest.mBio. 2024 Oct 16;15(10):e0024024. doi: 10.1128/mbio.00240-24. Epub 2024 Sep 13. mBio. 2024. PMID: 39269169 Free PMC article.
-
HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination.Oncogene. 2007 Jan 25;26(4):477-86. doi: 10.1038/sj.onc.1209831. Epub 2006 Sep 18. Oncogene. 2007. PMID: 16983346
-
Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair.Virol J. 2008 Jan 22;5:11. doi: 10.1186/1743-422X-5-11. Virol J. 2008. PMID: 18211700 Free PMC article.
-
Ataxia-telangiectasia and related diseases.Neuromolecular Med. 2006;8(4):495-511. doi: 10.1385/NMM:8:4:495. Neuromolecular Med. 2006. PMID: 17028372 Review.
-
DNA repair in HIV-1 infection: a case for inhibitors of cellular co-factors?Curr HIV Res. 2006 Oct;4(4):411-21. doi: 10.2174/157016206778560027. Curr HIV Res. 2006. PMID: 17073616 Review.
Cited by
-
An Overview of Human Immunodeficiency Virus Type 1-Associated Common Neurological Complications: Does Aging Pose a Challenge?J Alzheimers Dis. 2017;60(s1):S169-S193. doi: 10.3233/JAD-170473. J Alzheimers Dis. 2017. PMID: 28800335 Free PMC article. Review.
-
HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production.Redox Biol. 2018 May;15:97-108. doi: 10.1016/j.redox.2017.11.024. Epub 2017 Dec 7. Redox Biol. 2018. PMID: 29220699 Free PMC article.
-
Therapeutic doses of irradiation activate viral transcription and induce apoptosis in HIV-1 infected cells.Virology. 2015 Nov;485:1-15. doi: 10.1016/j.virol.2015.06.021. Epub 2015 Jul 14. Virology. 2015. PMID: 26184775 Free PMC article.
-
Improved Functionality of Integration-Deficient Lentiviral Vectors (IDLVs) by the Inclusion of IS2 Protein Docks.Pharmaceutics. 2021 Aug 6;13(8):1217. doi: 10.3390/pharmaceutics13081217. Pharmaceutics. 2021. PMID: 34452178 Free PMC article.
-
Lentiviral Vectors for Ocular Gene Therapy.Pharmaceutics. 2022 Jul 31;14(8):1605. doi: 10.3390/pharmaceutics14081605. Pharmaceutics. 2022. PMID: 36015231 Free PMC article. Review.
References
-
- Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
