

Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma
- PMID: 23435205
- PMCID: PMC3646862
- DOI: 10.4161/cc.24050
Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma
Abstract
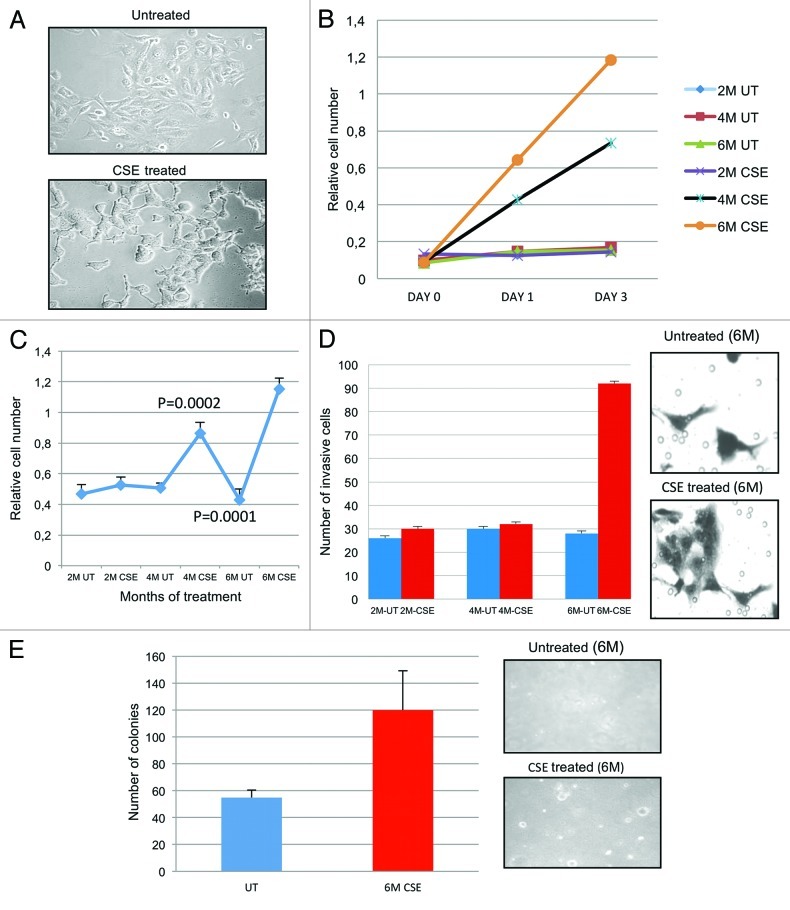
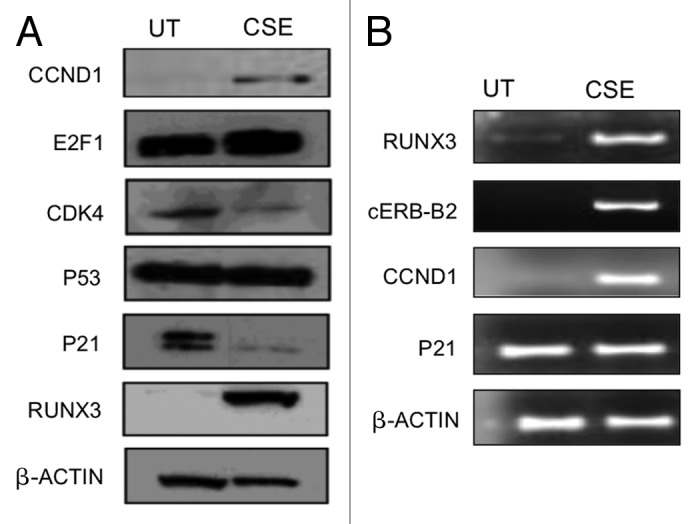
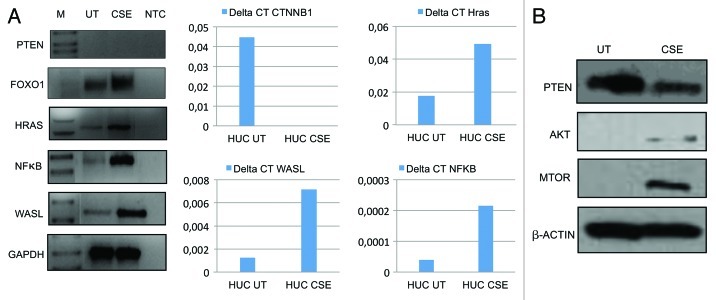
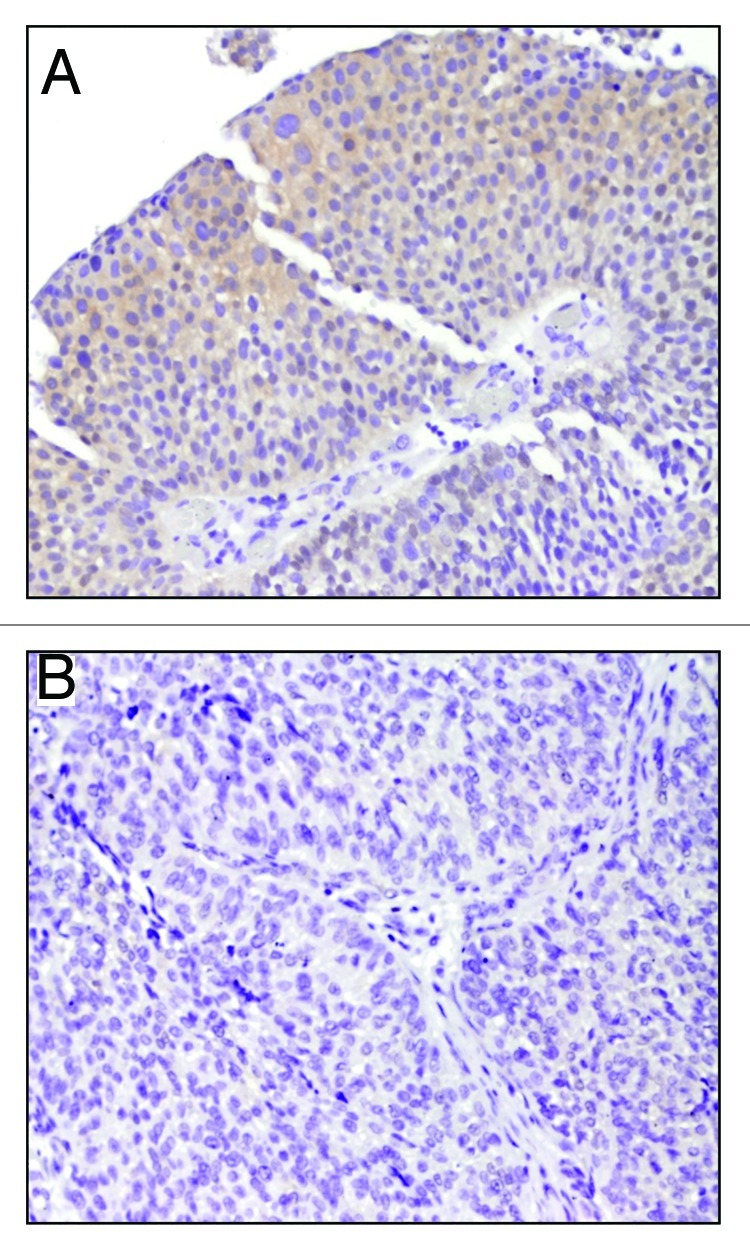
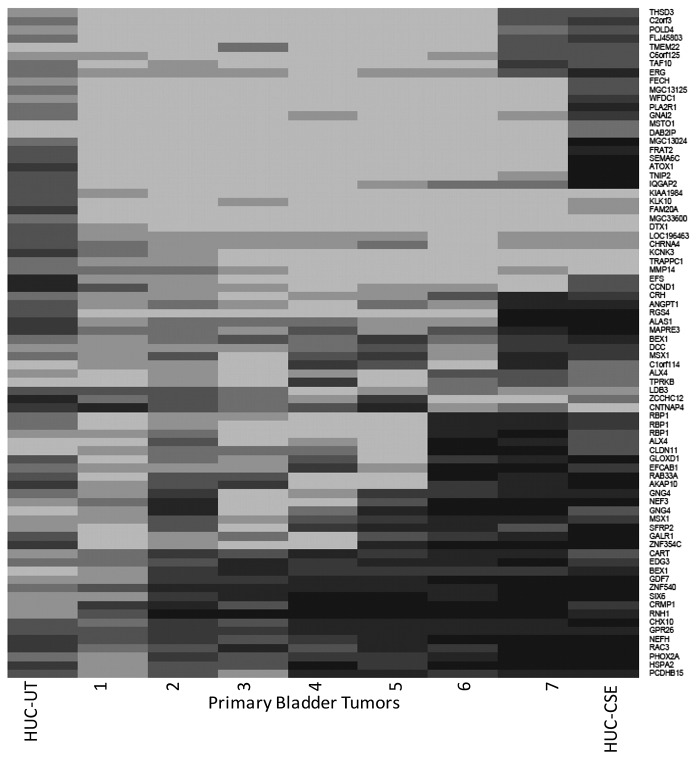
Urothelial cell carcinoma (UCC) is the second most common genitourinary malignant disease in the USA, and tobacco smoking is the major known risk factor for UCC development. Exposure to carcinogens, such as those contained in tobacco smoke, is known to directly or indirectly damage DNA, causing mutations, chromosomal deletion events and epigenetic alterations in UCC. Molecular studies have shown that chromosome 9 alterations and P53, RAS, RB and PTEN mutations are among the most frequent events in UCC. Recent studies suggested that continuous tobacco carcinogen exposure drives and enhances the selection of epigenetically altered cells in UCC, predominantly in the invasive form of the disease. However, the sequence of molecular events that leads to UCC after exposure to tobacco smoke is not well understood. To elucidate molecular events that lead to UCC oncogenesis and progression after tobacco exposure, we developed an in vitro cellular model for smoking-induced UCC. SV-40 immortalized normal HUC1 human bladder epithelial cells were continuously exposed to 0.1% cigarette smoke extract (CSE) until transformation occurred. Morphological alterations and increased cell proliferation of non-malignant urothelial cells were observed after 4 months (mo) of treatment with CSE. Anchorage-independent growth assessed by soft agar assay and increase in the migratory and invasive potential was observed in urothelial cells after 6 mo of CSE treatment. By performing a PCR mRNA expression array specific to the PI3K-AKT pathway, we found that 26 genes were upregulated and 22 genes were downregulated after 6 mo of CSE exposure of HUC1 cells. Among the altered genes, PTEN, FOXO1, MAPK1 and PDK1 were downregulated in the transformed cells, while AKT1, AKT2, HRAS, RAC1 were upregulated. Validation by RT-PCR and western blot analysis was then performed. Furthermore, genome-wide methylation analysis revealed MCAM, DCC and HIC1 are hypermethylated in CSE-treated urothelial cells when compared with non-CSE exposed cells. The methylation status of these genes was validated using quantitative methylation-specific PCR (QMSP), confirming an increase in methylation of CSE-treated urothelial cells compared to untreated controls. Therefore, our findings suggest that a tobacco signature could emerge from distinctive patterns of genetic and epigenetic alterations and can be identified using an in vitro cellular model for the development of smoking-induced cancer.
Keywords: Urothelial cell carcinoma; bladder cancer; cigarette smoke extract; epigenetics; in vitro transformation; smoking.
Figures
Comment in
-
Discovering smoking-related pathway alterations in urothelial cell carcinoma pathogenesis.Cell Cycle. 2013 May 15;12(10):1483. doi: 10.4161/cc.24852. Epub 2013 Apr 29. Cell Cycle. 2013. PMID: 23652923 Free PMC article. No abstract available.
References
-
- Wang YH, Yeh SD, Wu MM, Liu CT, Shen CH, Shen KH, et al. Comparing the joint effect of arsenic exposure, cigarette smoking and risk genotypes of vascular endothelial growth factor on upper urinary tract urothelial carcinoma and bladder cancer. J Hazard Mater. 2012 doi: 10.1016/j.jhazmat.2012.08.056. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous