

Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse
- PMID: 23436904
- PMCID: PMC3675825
- DOI: 10.1074/mcp.M112.024919
Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse
Abstract
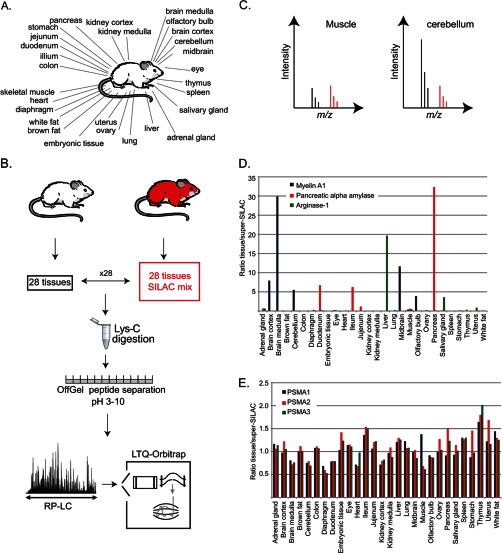
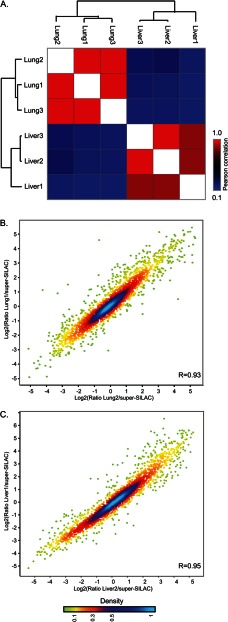
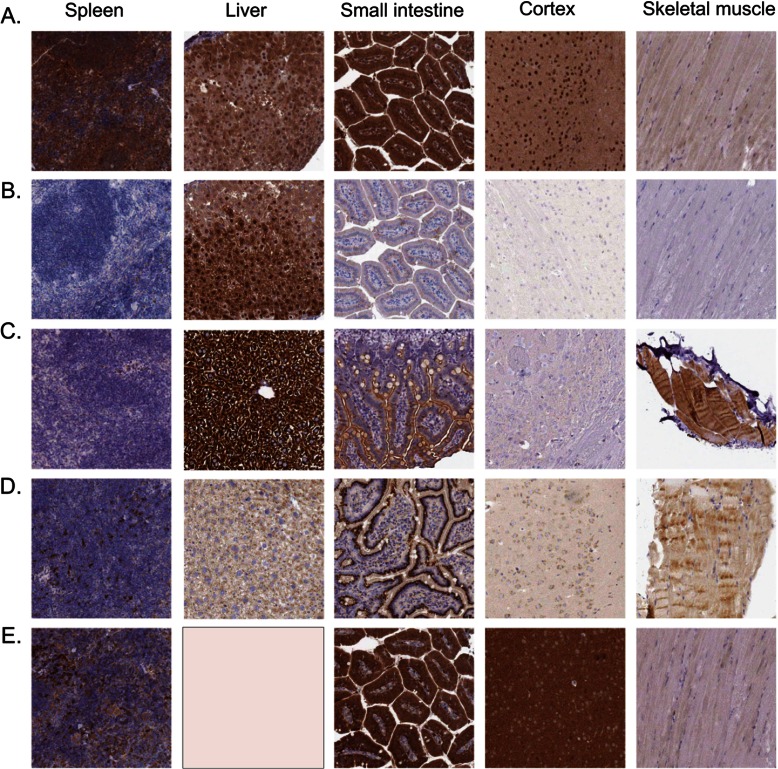
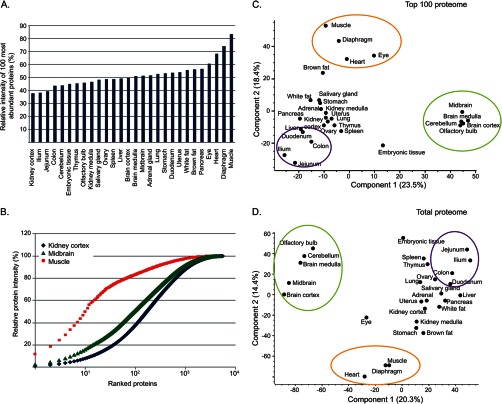
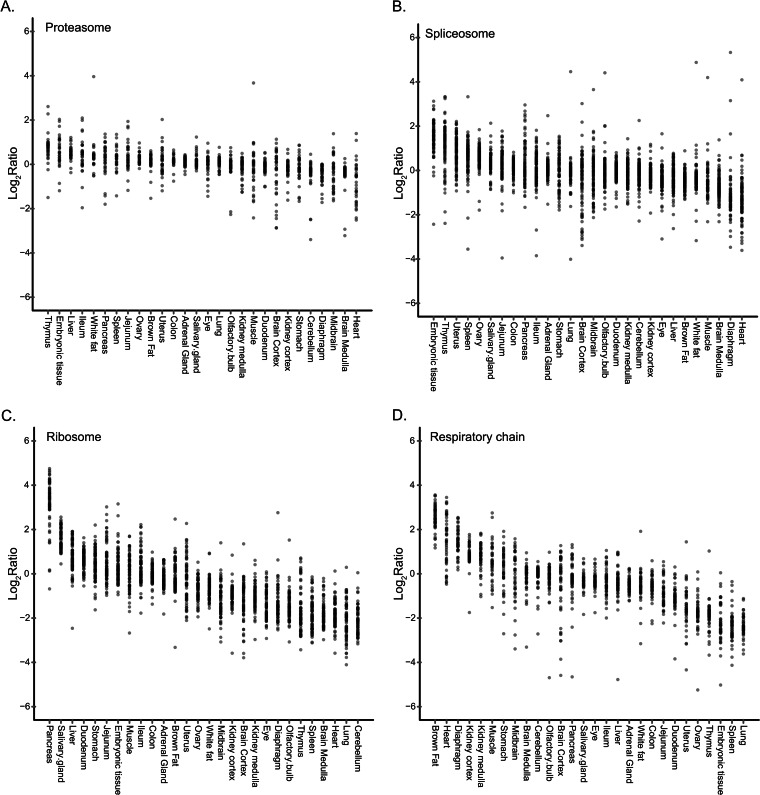
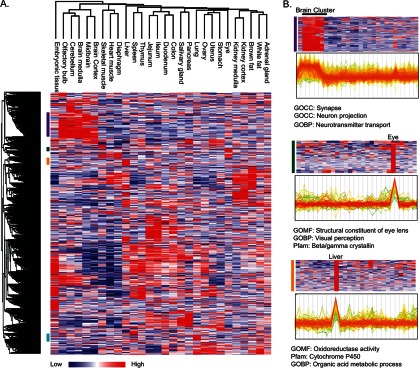
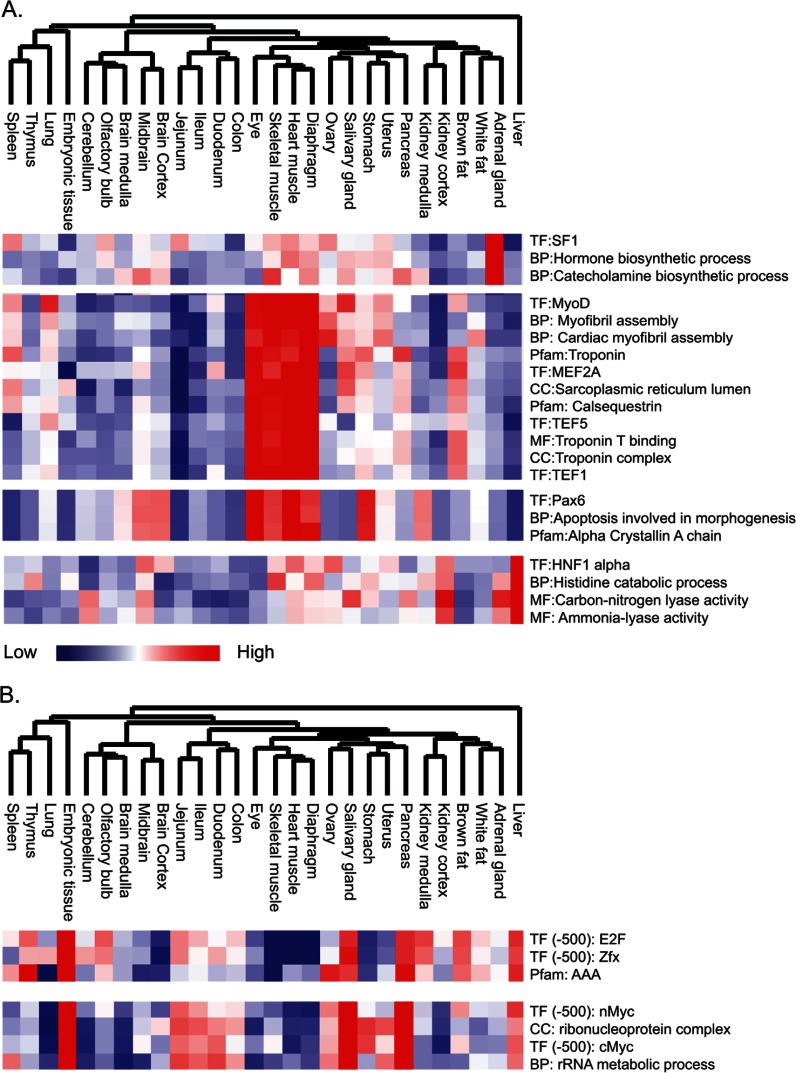
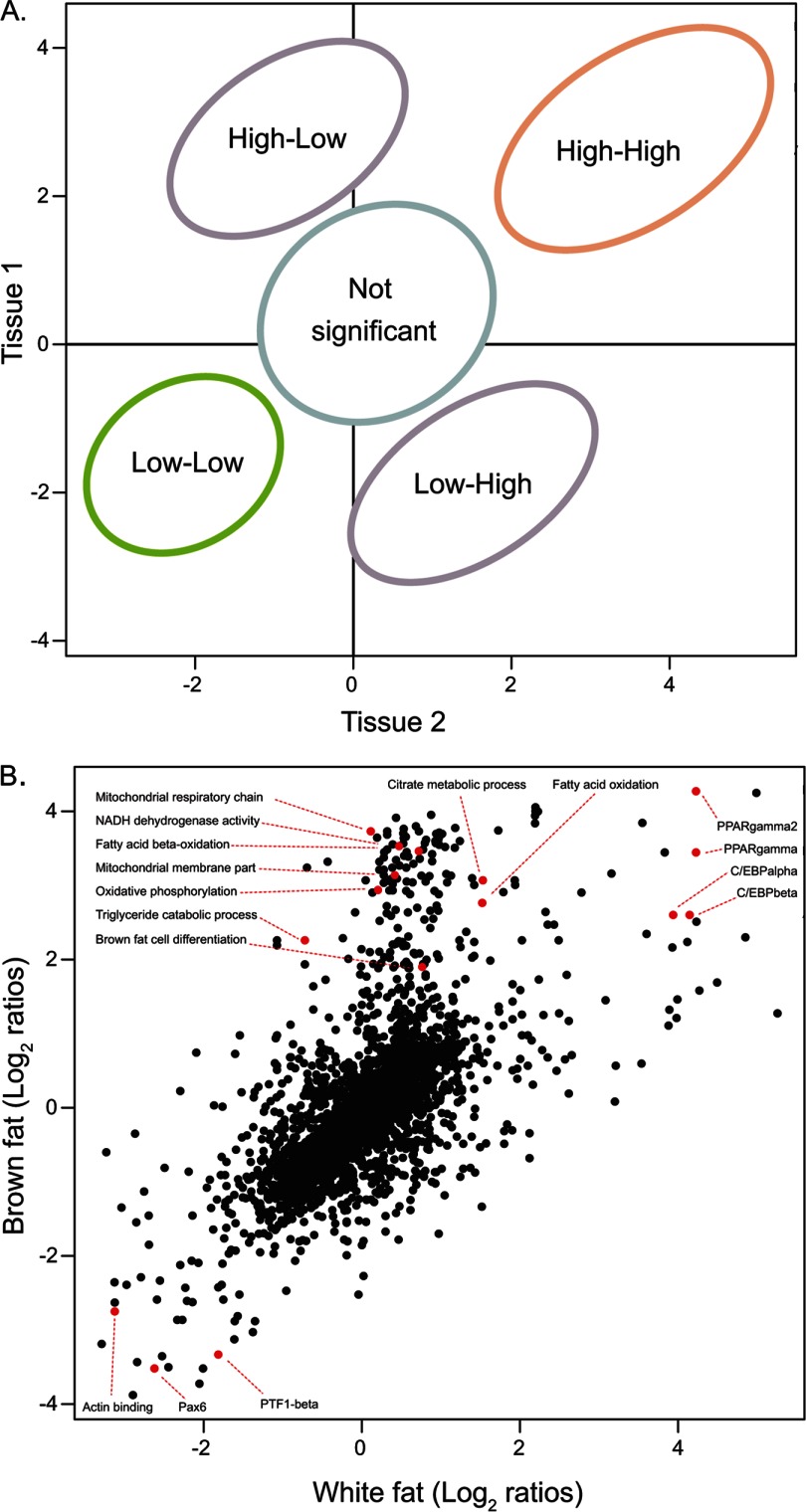
Identifying the building blocks of mammalian tissues is a precondition for understanding their function. In particular, global and quantitative analysis of the proteome of mammalian tissues would point to tissue-specific mechanisms and place the function of each protein in a whole-organism perspective. We performed proteomic analyses of 28 mouse tissues using high-resolution mass spectrometry and used a mix of mouse tissues labeled via stable isotope labeling with amino acids in cell culture as a "spike-in" internal standard for accurate protein quantification across these tissues. We identified a total of 7,349 proteins and quantified 6,974 of them. Bioinformatic data analysis showed that physiologically related tissues clustered together and that highly expressed proteins represented the characteristic tissue functions. Tissue specialization was reflected prominently in the proteomic profiles and is apparent already in their hundred most abundant proteins. The proportion of strictly tissue-specific proteins appeared to be small. However, even proteins with household functions, such as those in ribosomes and spliceosomes, can have dramatic expression differences among tissues. We describe a computational framework with which to correlate proteome profiles with physiological functions of the tissue. Our data will be useful to the broad scientific community as an initial atlas of protein expression of a mammalian species.
Figures
References
-
- Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
