

Patterns of prokaryotic lateral gene transfers affecting parasitic microbial eukaryotes
- PMID: 23442822
- PMCID: PMC4053834
- DOI: 10.1186/gb-2013-14-2-r19
Patterns of prokaryotic lateral gene transfers affecting parasitic microbial eukaryotes
Abstract
Background: The influence of lateral gene transfer on gene origins and biology in eukaryotes is poorly understood compared with those of prokaryotes. A number of independent investigations focusing on specific genes, individual genomes, or specific functional categories from various eukaryotes have indicated that lateral gene transfer does indeed affect eukaryotic genomes. However, the lack of common methodology and criteria in these studies makes it difficult to assess the general importance and influence of lateral gene transfer on eukaryotic genome evolution.
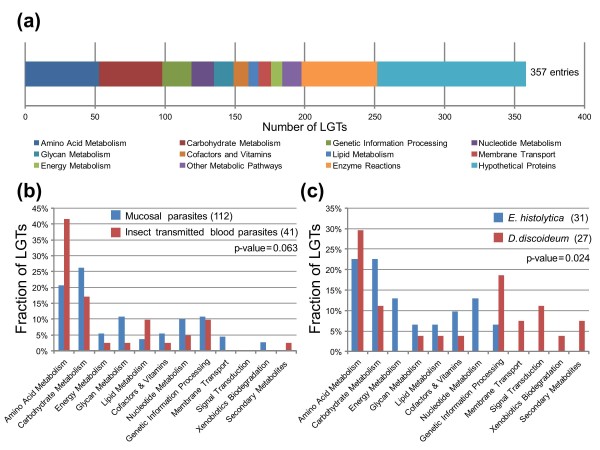
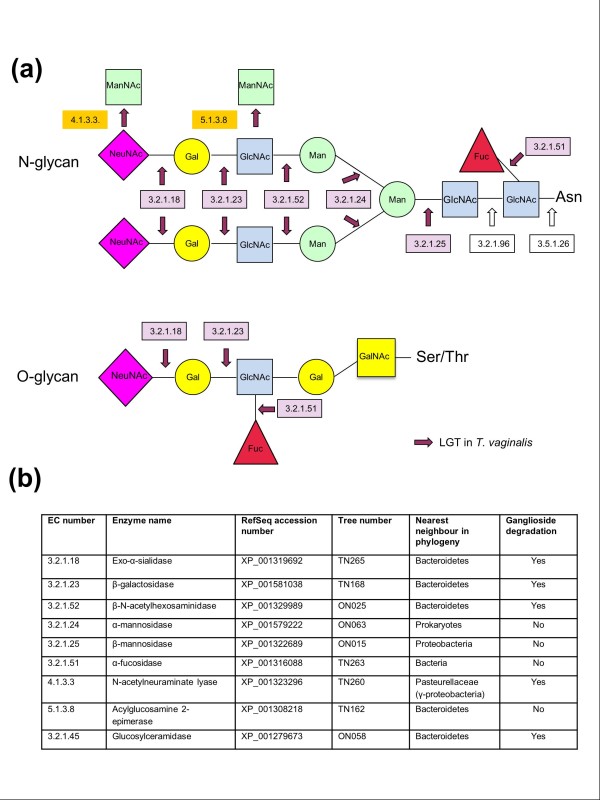
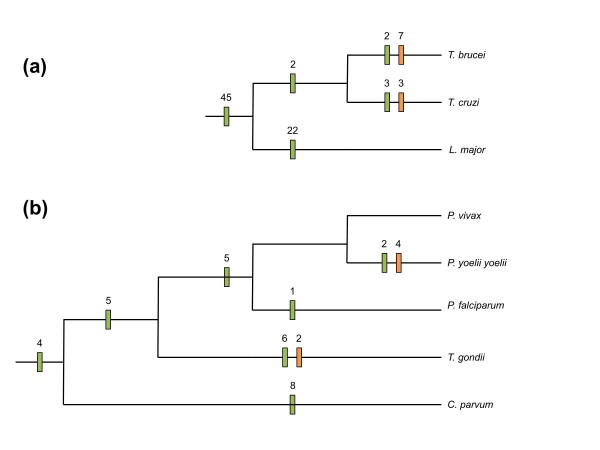
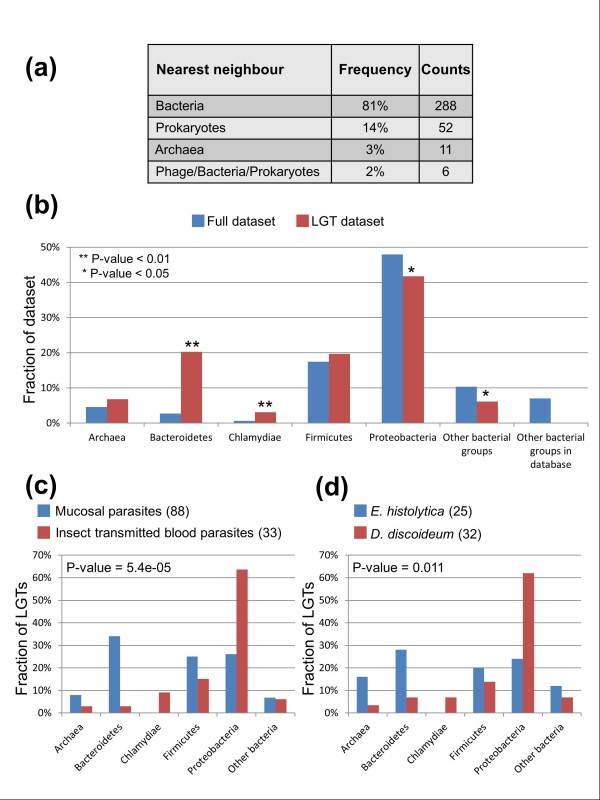
Results: We used a phylogenomic approach to systematically investigate lateral gene transfer affecting the proteomes of thirteen, mainly parasitic, microbial eukaryotes, representing four of the six eukaryotic super-groups. All of the genomes investigated have been significantly affected by prokaryote-to-eukaryote lateral gene transfers, dramatically affecting the enzymes of core pathways, particularly amino acid and sugar metabolism, but also providing new genes of potential adaptive significance in the life of parasites. A broad range of prokaryotic donors is involved in such transfers, but there is clear and significant enrichment for bacterial groups that share the same habitats, including the human microbiota, as the parasites investigated.
Conclusions: Our data show that ecology and lifestyle strongly influence gene origins and opportunities for gene transfer and reveal that, although the outlines of the core eukaryotic metabolism are conserved among lineages, the genes making up those pathways can have very different origins in different eukaryotes. Thus, from the perspective of the effects of lateral gene transfer on individual gene ancestries in different lineages, eukaryotic metabolism appears to be chimeric.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
