

Modeling the assembly of the multiple domains of α-actinin-4 and its role in actin cross-linking
- PMID: 23442921
- PMCID: PMC3566466
- DOI: 10.1016/j.bpj.2012.12.003
Modeling the assembly of the multiple domains of α-actinin-4 and its role in actin cross-linking
Abstract
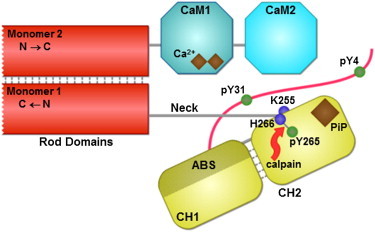
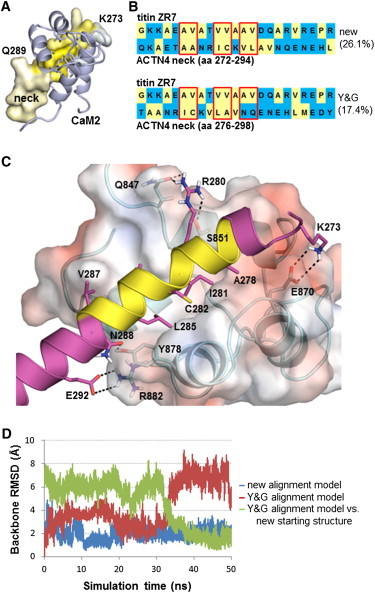
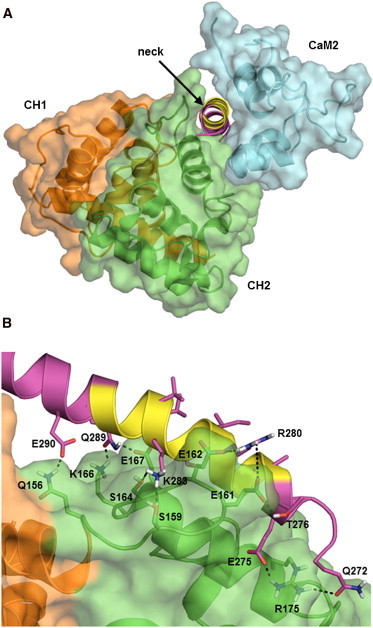
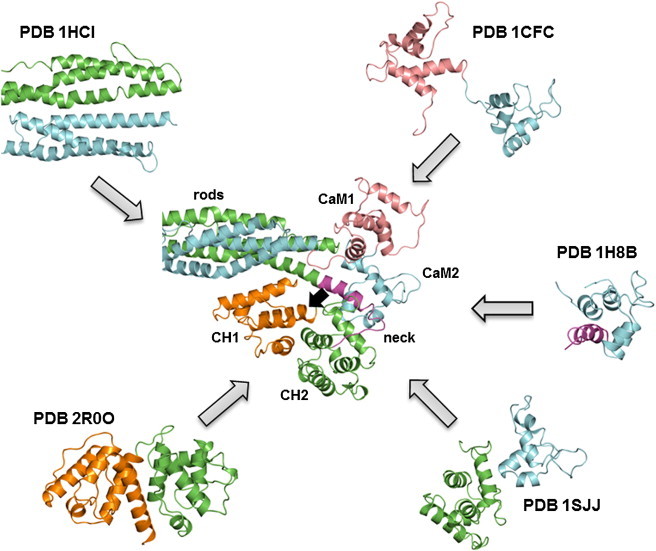
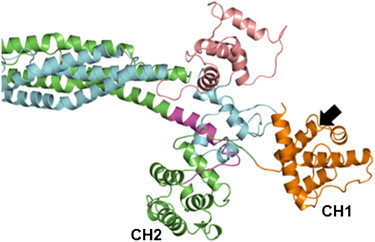
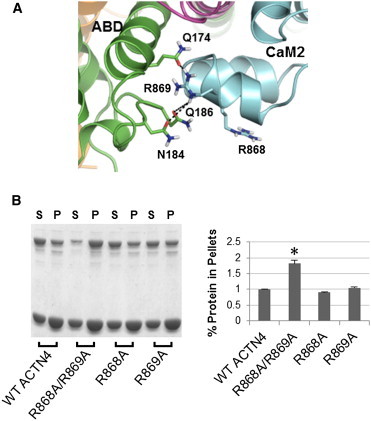
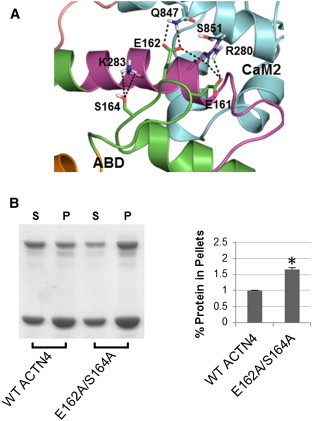
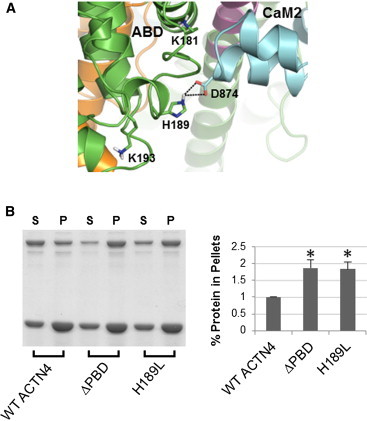
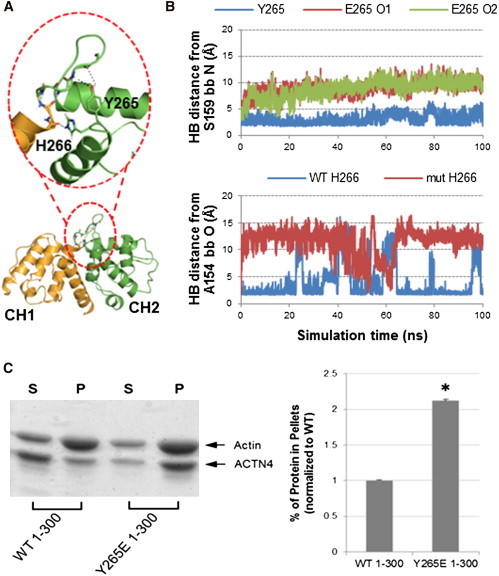
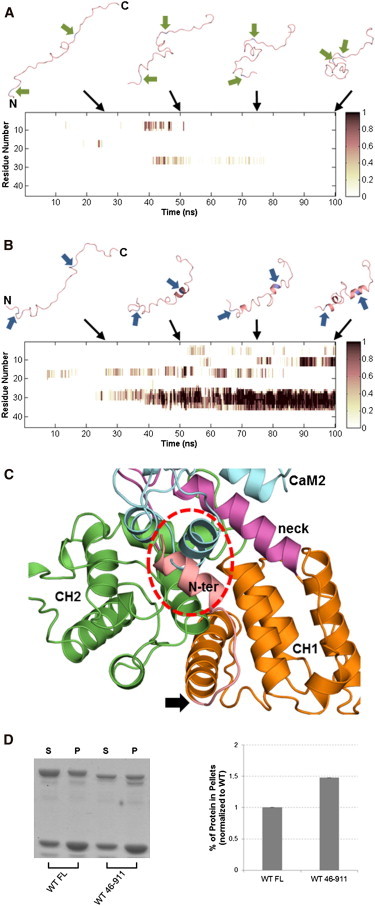
The assembly of proteins into multidomain complexes is critical for their function. In eukaryotic nonmuscle cells, regulation of the homodimeric actin cross-linking protein α-actinin-4 (ACTN4) during cell migration involves signaling receptors with intrinsic tyrosine kinase activity, yet the underlying molecular mechanisms are poorly understood. As a first step to address the latter, we validate here an atomic model for the ACTN4 end region, which corresponds to a ternary complex between the N-terminal actin-binding domain (ABD) and an adjacent helical neck region of one monomer, and the C-terminal calmodulin-like domain of the opposite antiparallel monomer. Mutagenesis experiments designed to disrupt this ternary complex confirm that its formation reduces binding to F-actin. Molecular dynamics simulations show that the phosphomimic mutation Y265E increases actin binding by breaking several interactions that tether the two calponin homology domains into a closed ABD conformation. Simulations also show a disorder-to-order transition in the double phosphomimic mutant Y4E/Y31E of the 45-residue ACTN4 N-terminal region, which can inhibit actin binding by latching both calponin homology domains more tightly. Collectively, these studies provide a starting point for understanding the role of external cues in regulating ACTN4, with different phenotypes resulting from changes in the multidomain assembly of the protein.
Copyright © 2013 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Dynamic Regulation of α-Actinin's Calponin Homology Domains on F-Actin.Biophys J. 2016 Mar 29;110(6):1444-55. doi: 10.1016/j.bpj.2016.02.024. Biophys J. 2016. PMID: 27028653 Free PMC article.
-
Phosphorylation of alpha-actinin 4 upon epidermal growth factor exposure regulates its interaction with actin.J Biol Chem. 2010 Jan 22;285(4):2591-600. doi: 10.1074/jbc.M109.035790. Epub 2009 Nov 17. J Biol Chem. 2010. PMID: 19920151 Free PMC article.
-
The crystal structure of the actin binding domain from alpha-actinin in its closed conformation: structural insight into phospholipid regulation of alpha-actinin.J Mol Biol. 2005 Apr 22;348(1):151-65. doi: 10.1016/j.jmb.2005.01.002. J Mol Biol. 2005. PMID: 15808860
-
Molecular biology of actin binding proteins: evidence for a common structural domain in the F-actin binding sites of gelsolin and alpha-actinin.J Cell Sci Suppl. 1991;14:91-4. doi: 10.1242/jcs.1991.supplement_14.19. J Cell Sci Suppl. 1991. PMID: 1653252 Review.
-
Novel structural insights into F-actin-binding and novel functions of calponin homology domains.Curr Opin Struct Biol. 2008 Dec;18(6):702-8. doi: 10.1016/j.sbi.2008.10.003. Epub 2008 Nov 13. Curr Opin Struct Biol. 2008. PMID: 18952167 Review.
Cited by
-
Synaptopodin couples epithelial contractility to α-actinin-4-dependent junction maturation.J Cell Biol. 2015 Oct 26;211(2):407-34. doi: 10.1083/jcb.201412003. J Cell Biol. 2015. PMID: 26504173 Free PMC article.
-
Postmitotic expansion of cell nuclei requires nuclear actin filament bundling by α-actinin 4.EMBO Rep. 2020 Nov 5;21(11):e50758. doi: 10.15252/embr.202050758. Epub 2020 Sep 22. EMBO Rep. 2020. PMID: 32959960 Free PMC article.
-
The Importance of Alpha-Actinin Proteins in Platelet Formation and Function, and Their Causative Role in Congenital Macrothrombocytopenia.Int J Mol Sci. 2021 Aug 29;22(17):9363. doi: 10.3390/ijms22179363. Int J Mol Sci. 2021. PMID: 34502272 Free PMC article. Review.
-
Tandem phosphorylation within an intrinsically disordered region regulates ACTN4 function.Sci Signal. 2015 May 26;8(378):ra51. doi: 10.1126/scisignal.aaa1977. Sci Signal. 2015. PMID: 26012634 Free PMC article.
-
Structure and calcium-binding studies of calmodulin-like domain of human non-muscle α-actinin-1.Sci Rep. 2016 Jun 7;6:27383. doi: 10.1038/srep27383. Sci Rep. 2016. PMID: 27272015 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
