

BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition
- PMID: 23447565
- PMCID: PMC3600451
- DOI: 10.1073/pnas.1205575110
BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition
Abstract
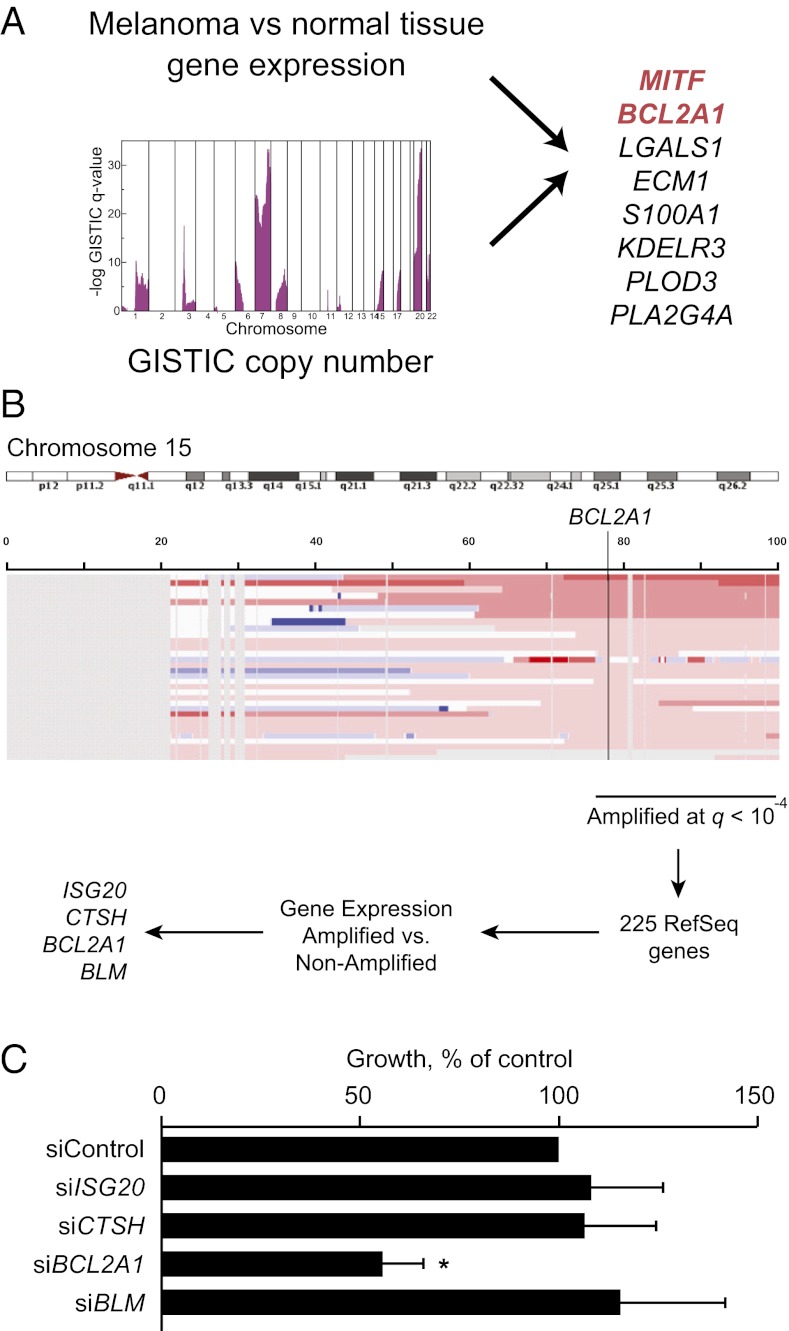
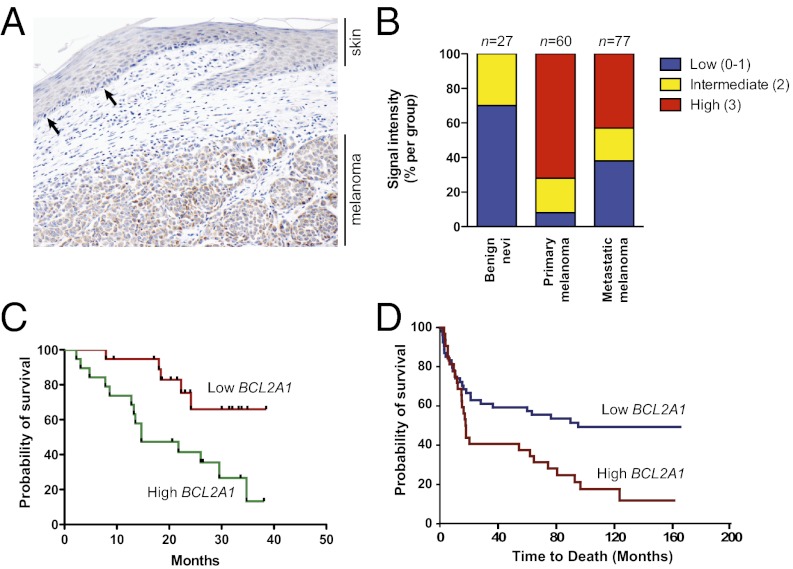
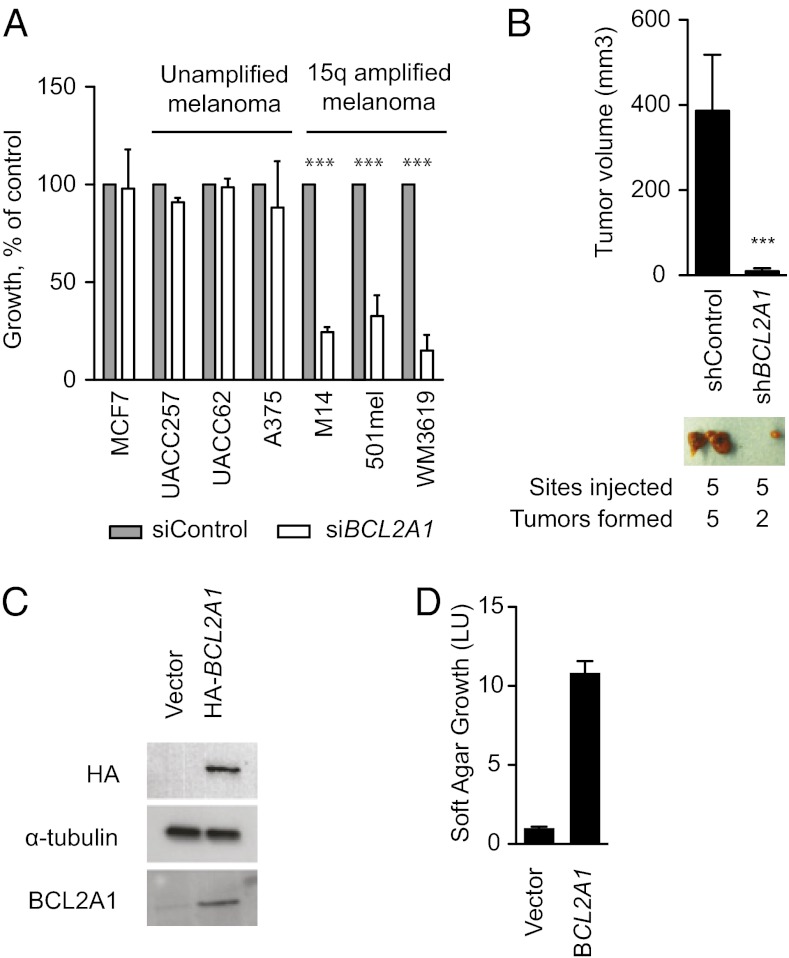
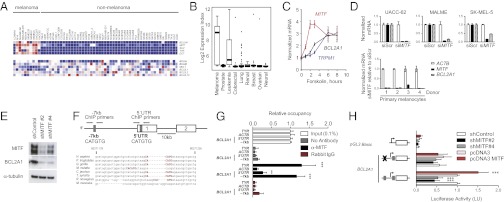
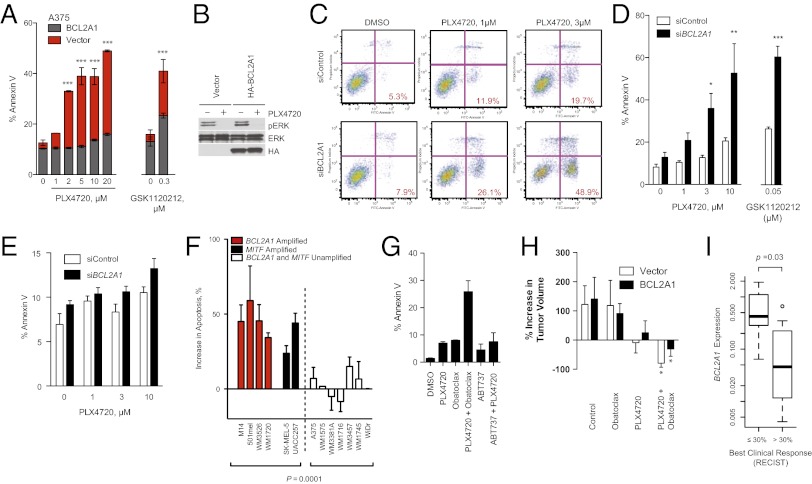
Although targeting oncogenic mutations in the BRAF serine/threonine kinase with small molecule inhibitors can lead to significant clinical responses in melanoma, it fails to eradicate tumors in nearly all patients. Successful therapy will be aided by identification of intrinsic mechanisms that protect tumor cells from death. Here, we used a bioinformatics approach to identify drug-able, "driver" oncogenes restricted to tumor versus normal tissues. Applying this method to 88 short-term melanoma cell cultures, we show that the antiapoptotic BCL2 family member BCL2A1 is recurrently amplified in ∼30% of melanomas and is necessary for melanoma growth. BCL2A1 overexpression also promotes melanomagenesis of BRAF-immortalized melanocytes. We find that high-level expression of BCL2A1 is restricted to melanoma due to direct transcriptional control by the melanoma oncogene MITF. Although BRAF inhibitors lead to cell cycle arrest and modest apoptosis, we find that apoptosis is significantly enhanced by suppression of BCL2A1 in melanomas with BCL2A1 or MITF amplification. Moreover, we find that BCL2A1 expression is associated with poorer clinical responses to BRAF pathway inhibitors in melanoma patients. Cotreatment of melanomas with BRAF inhibitors and obatoclax, an inhibitor of BCL2A1 and other BCL2 family members, overcomes intrinsic resistance to BRAF inhibitors in BCL2A1-amplified cells in vitro and in vivo. These studies identify MITF-BCL2A1 as a lineage-specific oncogenic pathway in melanoma and underscore its role for improved response to BRAF-directed therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Improving apoptotic responses to targeted therapy.Oncotarget. 2013 Sep;4(9):1331. doi: 10.18632/oncotarget.1261. Oncotarget. 2013. PMID: 23934756 Free PMC article. No abstract available.
-
Evolution of resistance to thyroid cancer therapy.Aging (Albany NY). 2016 Aug;8(8):1576-7. doi: 10.18632/aging.101030. Aging (Albany NY). 2016. PMID: 27575377 Free PMC article. No abstract available.
-
In Reply.Anesthesiology. 2017 Jul;127(1):202-203. doi: 10.1097/ALN.0000000000001692. Anesthesiology. 2017. PMID: 28632626 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
