

Genetic variants and their interactions in disease risk prediction - machine learning and network perspectives
- PMID: 23448398
- PMCID: PMC3606427
- DOI: 10.1186/1756-0381-6-5
Genetic variants and their interactions in disease risk prediction - machine learning and network perspectives
Abstract
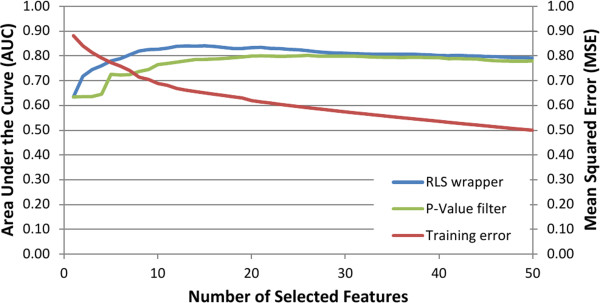
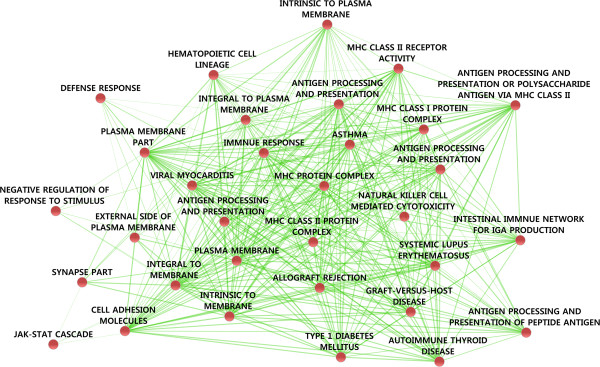
A central challenge in systems biology and medical genetics is to understand how interactions among genetic loci contribute to complex phenotypic traits and human diseases. While most studies have so far relied on statistical modeling and association testing procedures, machine learning and predictive modeling approaches are increasingly being applied to mining genotype-phenotype relationships, also among those associations that do not necessarily meet statistical significance at the level of individual variants, yet still contributing to the combined predictive power at the level of variant panels. Network-based analysis of genetic variants and their interaction partners is another emerging trend by which to explore how sub-network level features contribute to complex disease processes and related phenotypes. In this review, we describe the basic concepts and algorithms behind machine learning-based genetic feature selection approaches, their potential benefits and limitations in genome-wide setting, and how physical or genetic interaction networks could be used as a priori information for providing improved predictive power and mechanistic insights into the disease networks. These developments are geared toward explaining a part of the missing heritability, and when combined with individual genomic profiling, such systems medicine approaches may also provide a principled means for tailoring personalized treatment strategies in the future.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
